
Many multiscale modeling problems may be gainfully addressed as reducing degrees of freedom in nonlinear systems of ordinary differential equations. For instance, a closed molecular dynamic assembly, a spatially discretized system of equations from continuum mechanics representing a strongly inhomogeneous, nonlinear and possibly dissipative material, or a granular assembly operating under Newton’s laws and obeying elastic-viscous/plastic inter-particle contact laws serve as important examples of such systems. The primary multiscale analysis question may now be phrased as follows: We do not wish to solve the entire system but instead would like to consider the evolution of only a selected number of degrees of freedom of the whole system. Given that the system is coupled, is it possible to devise a strategy, perhaps approximate, to achieve such a goal? In particular, we would like to deal with situations where there is no intrinsic separation of time-scales available nor any small parameters for asymptotic techniques — e.g. flows on the ‘attractors’ of a dissipative dynamical system like the Lorenz system. We use a method of reduction involving converting the underlying autonomous ODE ‘fine’ system to a system of first-order PDE, the latter essentially representing (local) invariant manifolds in fine phase space. The idea goes back to Jacobi, and resurfaced in dynamical systems work since the middle 1960s (Sacker). Some methods of implementing Inertial Manifold Theory (IMT, Temam and co-workers) also employ the broad idea. It turns out that the type of solution one seeks to the PDE system is crucial in determining the practical success of the scheme with respect to model reduction. For example, it is known that the maximal reduction afforded by (IMT) is bounded below by the dimension of the ‘attractor.’ A practical consequence is that if the dimension of the attractor is large (e.g. Navier Stokes equations), then the reduced theory still contains a very large number of degrees of freedom. To us, this appears to be a direct consequence of requiring an autonomous (state-dependent) coarse theory that translates to seeking a solution of the system of PDE mentioned before as a graph over the entire coarse space. If this condition is relaxed, then it is possible to populate those regions of fine phase space, where (segments of) trajectories to be coarse-grained exist, by low dimensional, local invariant manifolds. A closed low dimensional coarse theory can now be set up whose solutions, essentially, ride this pre-computed set of local invariant manifolds of the fine theory, thus yielding self-consistency to coarse response. An important physical consequence is that the coarse theory in the case of such drastic reduction is, more often than not, history-dependent, i.e. produces self-intersecting trajectories in phase space; however, this history dependence is completely described by our method. Depending upon how energy in the coarse theory is defined, these ideas also demonstrate non-conservative coarse behavior arising from conservative fine systems. We have devised an algorithm based on the above idea that works reasonably on model problems. Its effectiveness in model reduction for the Lorenz system (chaotic behavior) as well as a nonlinear Hamiltonian system (periodic behavior) will be demonstrated. Time — permitting a few results from field dislocation mechanics will be demonstrated showing the emergence of microstructure at a very fine scale — coarse-graining such a theory to obtain a macroscopic theory of crystal plasticity is one of the goals of the model reduction method mentioned above. This is joint work with Aarti Sawant.
Pressing multiscale modeling problems in plasticity:
Developments in multiscale computational modeling lie at the heart of predictive, nonlinear solid mechanics. From the prediction of earthquakes to the dynamic response of polycrystals on to the inelastic response of single crystals at the submicron scale, success depends upon the extent to which coarse features of fine-scale behavior can be systematically and robustly computed without ad-hoc assumptions. This does not refer to constitutive response alone; coarse response of generally nonlinear fine systems could generally be governed by different governing balance laws that need to be discovered via coarse-graining or homogenization.
3-D discrete dislocation methodology provides a sound basis for the description of plasticity at the overall scale of microns (e.g. Amodeo and Ghoneim, 1990; Kubin et al., 1992; van der Giessen and Needleman, 1995). Field dislocation mechanics (Acharya, 2004) is developing methodology that allows the incorporation of nonlinear crystal elasticity, inertia, and the effects of geometric nonlinearity in dislocation mechanics. Both of the above techniques require large calculations at the scale of microns. If somehow, these calculations could be reduced in complexity by appropriately designed methods for model reduction in nonlinear systems, then it is conceivable that many such micron scale units can be coupled to represent a bigger continuum.
Similarly, if the computational burden in multiphase single crystal units deforming inhomogeneously may be reduced, then the coarse behavior of such units can be coupled to model material systems in geometries and under loading representative of important technological systems, e.g. single crystal, superalloy turbine blades (Busso et al., 2000). Bronkhorst et al. (1991) and Beaudoin et al. (1995) have developed approaches based on the Taylor model and the finite element method for polycrystalline response. The Taylor model fails to account for deformation inhomogeneities within grains, which has important effects. Multi-crystal aggregates have also been solved with computational single crystal plasticity theory but in these cases the case for a robust computational homogenization technique for strongly nonlinear systems becomes very strong due to the underlying cost.
There are groups in the US and EC working on such problems both in terms of developing relevant theories for small scale response as well as coarse-graining techniques for such models utilizing varied methods. Because of the extreme difficulty of the problem at hand and the sheer effort required to span the many scales involved, synergy and cross-fertilization of ideas between US and European researchers can be very beneficial in making progress on the above goal.
References:
It is well known that in polycrystalline metals, a substantial increase in strength and hardness can be obtained by reducing the grain size to the nanometer scale. These attributes have generated considerable interest in the use of nanocrystalline metallic materials (grain sizes less than .100 nm), for a wide variety of structural applications. Typically, relative to their microcrystalline counterparts, nanocrystalline metals exhibit a very high tensile strength, but at the expense of a much reduced tensile ductility. The limited ductility is of major concern. For example, while the ultimate tensile strength levels approach 1500MPa in electro-deposited nanocrystalline nickel, the ductility that can be obtained in this material is generally low and usually does not exceed 3%. Physical experiments and atomistic simulations reported in the literature, show that grain-boundary-related slip and separation phenomena begin to play an important role in the overall inelastic response of a polycrystalline material when the grain-size decreases to diameters under 100 nm, and dislocation activity within the grain interiors becomes more difficult. In order to model the effects of grain boundaries in polycrystalline materials we have coupled a crystal-plasticity model for the grain interiors with a new elastic-plastic grain- boundary interface model which accounts for both reversible elastic, as well irreversible inelastic sliding-separation deformations at the grain boundaries prior to failure. We have used this new computational capability to study the deformation and fracture response of nanocrystalline nickel. The results from the simulations capture the macroscopic experimentally-observed tensile stress-strain curves, and the dominant microstructural fracture mechanisms in this material. The macroscopically-observed nonlinearity in the stress-strain response is mainly due to the inelastic response of the grain boundaries. The stress concentrations at the tips of the distributed grain-boundary cracks, and at grain-boundary triple junctions, cause a limited amount of plastic deformation in the high-strength grain interiors. The competition of grain-boundary deformation with that in the grain interiors determines the observed macroscopic stress-strain response, and the overall ductility. In nanocrystalline nickel, the high yield strength of the grain interiors and relatively weaker grain-boundary interfaces account for the low ductility of this material in tension.
We have developed efficient computational codes based on the (orthogonal and non-orthogonal) Tight-Binding Molecular-Dynamics (TBMD) method including s,p,d basis set orbitals, electron correlations at the Hubbard-U level of approximation and Spin-Orbit interaction terms. We have used these codes successfully for investigating:
In this presentation, we demonstrate the efficiency of our codes in studying electronic, magnetic and transport properties of a wide class of nano-materials with significant technological importance. Emphasis is focused at the transport properties of some branched (Y- or T-shaped) SWCNs which show characteristic properties that allow them to be used as ballistic rectifiers or switches. Also the stability of various SiC nanotubes is demonstrated and finally it is shown that the encapsulation of TMAs within Si-based cage clusters leads to stable metal-encapsulated Si cage clusters (Si-cc) and Si-nanotubes (Si-NTs) the latter showing zero-energy conduction gap as their length becomes infinite. As it is demonstrated, the stabilization of these Si-based cages and tubes as well as their magnetic properties are strongly guided by a delicate interplay between the attainable symmetry of the system and the d-band filling of the encapsulated TMAs.
The work is supported by the EU GROWTH project AMMARE (contract number G5RD-CT-2001-00478).

Carbon nanotube T-junctions : Formation pathways and conductivity
Recent References
Views on EU-US Collaboration
Among the scientific issues I would like to refer to are:
Multiscale calculation seeks to improve computational efficiency by describing a physical system on a hierarchy of different levels. Ideally, the results of such calculations should approach those of the most reliable member of the hierarchy in a well-defined limit. However, the necessarily approximate nature of the coarser levels of the hierarchy makes this ideal elusive. This talk presents three examples of approaches which achieve ideal in the context of ab initio density-function theory calculations: (1) Use of multiresolution analysis (wavelet theory) to provide the first new electronic structure method to compete directly with full-potential linear augmented plane wave (FP-LAPW) calculations in terms of accuracy, but with far fewer and more transparent adjustable computational parameters; (2) Linkage of atomistic potential models with ab initio density functional theory calculations to compute exact density-functional thermal averages at greatly accelerated rates; (3) Introduction of a new, exact density-functional theorem allowing the rigorous separation of a system from its environment with application to the behavior of material and chemical systems in solution.
Silicon nitride (Si3N4) films on silicon substrates have a wide variety of applications in electronics and photovoltaics.1 Recent experiments by Kim and Yeom2 indicate that the thermally grown Si3N4 film on Si(111) has an atomically abrupt and defect-free interface. Their finding supports the model used in the molecular-dynamics simulations presented. In both applications, the interface can be subject to extreme environments and conditions causing strains, e.g., occurring at various strain rates. Atomistic simulations complement experimental research to gain fundamental understanding of possible failure mechanisms, which, in turn, enables the production of reliable components in microelectronics and photovoltaics applications. Silicon is modeled by the well-known Stillinger-Weber potential3 and was adapted to describe a silicon system that is expanded so that it perfectly matches silicon nitride. Bulk Si3N4 is modeled using a combination of two- and three-body interactions, which include charge transfer, electronic polarizability, and covalent bonding effects.4 The interface atoms are treated differently than those in the bulk to describe the bonding across the Si/Si3N4 interface. To account for all the structural correlations between silicon and silicon nitride, eight different components are used to model the silicon/silicon nitride system.5 The mechanical strength of the Si/Si3N4 interface was investigated by applying tensile stress parallel to the interface. Calculations of the Young's modulus of this particular interface showed that the value of 185.498 (� 0.29) GPa for the silicon/silicon nitride interface lies, as expected, comfortably between the Young's moduli of silicon and silicon nitride, respectively. At low strain rates, we found that when systems were stretched continuously, those that were stretched more quickly failed at higher strains. The failure mechanism was a crack in silicon nitride and plastic deformation in silicon. At the highest strain rate the stress is released through plastic deformation in silicon nitride, a qualitatively different failure mechanism compared to the fracture in silicon nitride at lower strain rates. A detailed analysis of the failure of the silicon/silicon nitride interface will be presented.
* Priya Vashishta developed the model for the silicon/silicon nitride interface during my stay as post-doc with him, Rajiv Kalia, and Aiichiro Nakano at LSU. The work presented here was supported in part by NASA, NSF, and a WVU Faculty Senate Grant.
References
Multi-scale Simulations of Interface Systems
Within the past couple of years, enormous progress has been made in developing and improving simulation approaches that couple multiple length scales.1 There are three main paths followed to link various length scales in order to solve complex materials problems. One path is serial: the simulation at the smaller scale yields results, such as material parameters, that are used as input for the simulation at the larger scale. Concurrent simulation approaches are presently done in two different ways: i) Simulations at different scales are integrated by introducing “handshake” regions where two types of approaches meet;2 ii) Derived scaling is applied by using an underlying fine-scale model for the entire system and removing degrees of freedom explicitly at scales up to the scale of interest.3
As classical interatomic potentials are already highly developed and capable of describing many phenomena, the concurrent hybrid finite-element/molecular-dynamics approach is adequate in many situations. However, well-studied situations that require a more refined treatment are, e.g., regions around crack tips where the breaking of bonds is most accurately described using quantum simulations. Another less-studied situation is that of interfaces between two materials. The binding of atoms across an interface differs very much from the one in a bulk sample. The most accurate model would therefore combine continuum simulations away from the interface, classical molecular-dynamics simulations when approaching the interface, and quantum calculations at the interface and in the direct vicinity of the interface (see figure).
Whereas the extension of the molecular-dynamics simulation method to larger length scales by the hybrid finite-element/molecular-dynamics approach is computationally highly efficient, the inclusion of quantum mechanical calculations typically requires the majority of the computational allocation and time.4
Alexander Shluger (University College London) was opening Friday.s session with his view of the necessity of multi-scale computational modeling of defect processes at oxide interfaces. My own presentation concluded at the very end of Friday.s session by discussing the importance of multi-scale modeling of interfaces as described above. Using a multi-scale approach we can study structural, mechanical, electrical, optical, and other materials properties. In discussions with various European and US participants in the NSF/EC workshop I have found that there is great interest in accurately modeling interface systems by combining the expertise of researchers who do not only want to bridge length- and time-scales but also the Atlantic.

Nanoscale and molecular computing promises to revolutionize computing by vastly increasing the processing speed and dramatically decreasing power requirements, enabling superfast image recognition and ultradense circuitry, as well as sensors with close to single-molecule detection limit. However, the experimental design of such devices is only at the beginning stages. Explicit and reliable modeling of such systems could greatly speed up the progress. However, the tools necessary for such modeling are only beginning to emerge. Focusing first on the electron transport problem in the device, it cannot be handled using standard quantum chemistry and electronic structure methods, because a working electronic device is an open system, with electrons entering and leaving the device through very long leads kept at fixed potentials. Several methods for treating such systems have been developed,1 but there are major questions concerning the reliability of the various techniques and their range of applicability. While the "gold standard" for validating theoretical approaches is comparison with experiment, such comparisons are exceedingly difficult at present, due to uncertainties in experimental measurements. Furthermore, a realistic device structure is quite complicated and consists of several active components, which all need to be faithfully modeled. An appropriate description would thus be inherently multiscale, accounting for the self-assembly of the molecular components, the electronic structure and the electron propagation through the nanodevice, and the coupling to the source and drain. The issues here are both a sufficiently accurate description of the electronic structure and electron transport (which may require the inclusion of many-body effects) and the ability to handle a large number of atoms, so that a realistic device structure is being studied. An accurate, multiscale description of the assembly process is also sorely needed, because in the low-resistance "ballistic" regime most of current loss occurs at contacts, which are poorly characterized at present. Understanding of this process would result in reliable models of contact structure, which would be used in simulations of electron flow and scattering at the contacts.

Another important aspect of future advances in nanoscale computing and sensing is the integration of the new devices with traditional semiconductor electronics. For example, new, exciting functionality could be obtained by integrating biomolecules and biology-inspired processing steps with Si electronics, which could result in new biosensors, as well as precise, molecular-level control over growth and processing of nanoscale devices. Although these prospects are clearly very exciting, the current understanding of key steps is clearly insufficient and multiscale modeling would be of great help here, especially if closely coupled to the experimental efforts. For example, if it was possible to precisely characterize the adsorption and subsequent reactions of key biomolecules on surfaces, processes could be devised that would self-assemble and grow the desired structures one molecular layer at a time, in a manner akin to atomic layer epitaxy,2 where crystal growth proceeds by deposition of single self-terminating monolayers, one at a time.
The identification and characterization of the surface structures may be best pursued by a combination of theoretical and experimental efforts. For example, optical techniques enable surface diagnostics during growth, even if ambient gas is present. The optical line shape can be calculated by electronic structure methods and good agreement between experiment and theory has enabled identification of several reconstruction patterns on semiconductor surfaces. However, it has also been discovered that the inclusion of many-body effects is essential and these effects are likely to be more pronounced for biomolecules, due to their large excitation energies. A theoretical investigation of optical excitations in biomolecular systems would thus be important, as would multiscale modeling of the various stages of the assembly, reactivity and growth of biomolecules on semiconductor surfaces.
Two modeling studies related to the production of AlN crystals via vapor transport will be discussed. In the first study a new, parameter-free first principles strategy is used that not only yields mole fractions of gas-phase species as a function of reactor conditions, but also identifies growth precursors based on their degree of saturation with respect to the growing crystal. The strategy predicts that Al and N2 are present in high relative concentrations, in agreement with available experimental measurements, but that N2 molecules are undersaturated with respect to the AlN crystal and therefore are unlikely growth precursors. Instead, Al N2, Al3N, and Al4N species, while in much smaller concentrations than N2, are predicted to be supersaturated and therefore are the main source of nitrogen contributing to AlN crystal growth, in stark contrast to assumptions made in prior modeling studies. In recent experiments it has been noted that AlN crystals deposited in BN crucibles tend to grow faster in the c direction with smooth (0001) facets compared to crystals grown in W or TaC crucibles. We propose that trace boron impurities arising from the crucible preferentially incorporate into steps in the AlN surface and lower the Schwoebel diffusion barrier, leading to enhanced step growth and decreased secondary nucleation. This proposal is supported by molecular modeling studies using classical potentials, which show that the strain energy associated with B substitution drives B impurities to steps, and that the resulting strong B-N bond to surface adsorbates can reduce the Schwoebel barrier.
*Funded by the Office of Naval Research through MURI contract N00014-01-1-0302.
The bridging between the mechanical behavior of an individual phase or crystal and that of a polycrystal remains a topic of major interest and is at the heart of homogenization schemes developed to predict the behavior of heterogeneous materials at different scales. Such schemes are based on the assumption that the mechanical behavior of individual constituents can lead to the description of the mechanical response of a macroscopic aggregate through either suitable interaction laws or a numerical averaging process of a representative volume element (RVE) of the microstructure. In this presentation, a multiscale constitutive framework recently proposed to describe the mechanical behavior of heterogeneous microstructures will be discussed. The framework enables explicit links between the dominant microstructural features at the microscopic and mesoscopic scales to be made with the macroscopic constitutive behavior of the material. The approach has been implemented into the finite element method and is used to examine the effect of the volume fraction of eutectic microstructures and precipitates on the mechanical behavior of a heterogeneous Ni-base single crystal and directionally solidified superalloys. The behavior of suitable RVEs is computed from actual digitized images of typical microstructures, and a multiscale crystallographic constitutive model is then formulated to describe the mechanical behavior of the homogenized material at the macroscale. The implications of using these types of multi-scale modeling capabilities within an industrial context and the potential for developing links with the atomistic scale will be discussed.
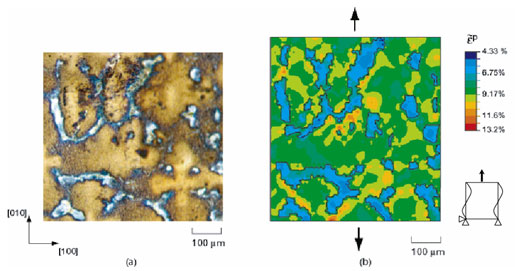
Homogenisation scheme applied to a typical heterogeneous single crystal superalloy: (a) RVE and (b) predicted contours of accumulated inelastic strain under [010] uniaxial loading
Recent References
In this talk I will present a novel approach, called First Principles String Molecular Dynamics, to find chemical reaction pathways in the context of First Principles Molecular Dynamics simulations. Applications to selected chemical reactions in condensed and gas phases will be used to illustrate the scheme. As found by previous investigators, current GGA approximations of the exchange-correlation functional, tend to underestimate reaction barriers. I will show that, in the case of the reactions studied here, a recently developed meta-GGA functional results in barrier heights that are in closer agreement with experiment and with calculations based on more accurate quantum mechanical methods. Finally, I will show that meta-GGA improves significantly DFT based predictions of melting transitions.
Point defects in silicon and silicon surfaces. Silicon based devices recover an enormous importance in material science. In the bulk material, the formation, diffusion and interaction of defects largely affect the electronic properties. Concerning surfaces, the comprehension of the phenomena of surface relaxation, reconstruction, passivation, and more generally of the adsorption of external elements, could support the interpretation of the processes of passivation, crystal epitaxial growth and the engineering of self-assembled layers. In the last few years, we studied the processes of migration and clustering of native point defects in silicon.1, 2 They act as intermediates for the growth of intrinsic extended defects, and determine many properties of the bulk material such as, for example, its behavior under irradiation. To investigate these systems we devised a two step strategy. First, we used a semiempirical approach (periodic Tight Binding Molecular Dynamics) to model the dynamical evolution of the defective crystal. Secondly, we selected representative instantaneous atomic configurations from the dynamical simulations to study the electronic properties of the defects. At this stage, first principles computations (Hartree-Fock) were performed on appropriate silicon cluster models with geometries derived from the simulations. We then analyzed how the bonding network and the atomic properties of the investigated systems evolve by performing a topological analysis of their electron density, within the formalism of the Quantum Theory of Atoms in Molecules.3 The combination of these techniques enabled us to describe processes involving hundreds of atoms at an atomistic level. The approach may be extended to study non intrinsic defects, which involve doping elements relevant for technological applications and industrial processes (for example hydrogen, boron, oxygen and metal impurities). As concerns silicon surfaces, we studied the systems which are more experimentally diffuse: clean and H-covered Si(111)(1x1);4 clean Si(111)(2x1);5 Si(100)(1x1):H; clean and H-covered Si(100)(2x1). We performed first principles computations (periodic Hartree-Fock and Density Functional) to optimize their geometry, and then we focused on the relations between structural and electronic rearrangements respect to the ideal crystal. We evaluated how the bonding and atomic properties of the surface atoms are related to the processes of relaxation, reconstruction and H covering. Analogously to the case of point defects, the combination between dynamical simulations and first principles computations could be very useful to model and understand quite complex phenomena occurring at the surface-vacuum interface. Moreover, the theoretical study of the electron density of regular surfaces is relevant in itself to establish a link with the recent experiments of surface X-ray scattering crystallography using synchrotron radiation facilities.
Thermoelectrics. Our research on thermoelectric materials was financed by the European Union under the Nanothermel Project. This involved research institutions and industrial partners from six different European countries. Thermoelectric devices may be used as power generators and as coolers. In this latter case, they reach an efficiency of about 10% of the Carnot limit, which is about three times smaller than for traditional coolers. This claims for significant improvements to be pursued on these devices. The kernel of a thermoelectric device is the two materials forming each element, and the very important point is to optimize and increase the material's figure of merit ZT (ZT=TS2s/k;T=absolute temperature; S Seebeck coefficient; s=electrical conductivity; k=thermal conductivity). Most of the factors determining ZT are critically dependent on the material electronic band structure (S, s, and the electronic contribution to k), and the focus of our research was to understand, at an atomistic level, how to modify the band structure to improve ZT. On the experimental side, S, s and k are tuned mainly by means of doping and nanostructuring, but the optimization strategy may vary from material to material. According to the work plan of the whole project, we had the task to perform first principle computations for supporting the characterization and the selection of the most promising thermoelectrics. More precisely, we studied the role of point defects in determining the thermoelectric properties, giving, if possible, indications on optimal doping elements and doping level. Secondly, we supported the interpretation of structural experiments, especially when these latter did non provide clear-cut answers. Third, we characterized the nature of the chemical interactions present in these materials within the formalism of the Quantum Theory of Atoms in Molecules. These general guidelines have been applied to the study of Skutterudites,6 inorganic Clathrates7 and Zinc-Antimonides.8 Additional information of these systems may be recovered by studying the role of point defects and nanostructuring in determining the lattice contribution to k.
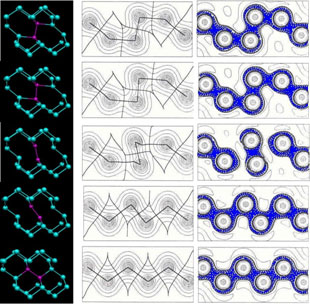
Fig. 1. Left panels: stick and ball representation of the annihilation of the bond defect (top) to form the ideal silicon crystal (bottom). This metastable structure was observed during a Tight Binding Molecular Dynamics simulation of the recombination between a vacancy and a self interstitial defect in silicon. Middle panels: contour lines of the electron density, obtained from cluster Hartree-Fock computations. Right panels: Laplacian of the electron density distribution; in the regions of electron density concentration, painted in blue, the Laplacian becomes large and negative. The study of the features of the electron density distribution allows to recover the evolution of bonding and atomic properties of the bond defect along the annihilation.

Fig. 2 Left panels: stick and balls representation of the building blocks of ß-Zinc-Antimonide, one of the most promising thermoelectric materials. From top to bottom: the ideal framework, containing twelve zinc atoms per cell; one zinc atom is then removed (A) and a zinc dimer (B, C) inserted in the cavity of the vacancy; the zinc dimer may induce a rearrangement in the surrounding zinc framework, involving the first zinc neighbor A'; bottom panel: the cavity of a single zinc cavity may host up to three zinc atoms (B, C and D). Right panels: density of states of these structures, as obtained from periodic Density Functional computations; the Fermi level is traced in red. Cells containing twelve zinc atoms are p-doped semiconductors (top panel); the insertion of an additional zinc atom (middle panels) completely fills the valence bands; the displacement of zinc induced by the dimer has no relevant effects on the electronic structure; further zinc insertion (bottom panel) correspond to n-doping.
References
The properties of matter at the nanoscale are quite different than their macroscopic counterparts. For example, optical excitations in porous silicon are strongly blue shifted from crystalline silicon owing to quantum confinement. I will illustrate some recent theoretical progress in developing numerical approaches to compute the optical and electronic properties of semiconductor materials whose physical dimensions are on the nanoscale. I will focus on real space methods for solving the electronic structure problem in this size regime.
Recent References
An intriguing and unexpected feature has recently been discovered during epitaxial growth of metal thin films on semiconductors. Instead of forming three-dimensional (3D) islands of various size as commonly observed for nonreactive interfaces, the metal atoms can arrange themselves into plateaus or islands of selective heights with flat tops and steep edges under certain growth conditions. This unusual behavior has been observed in quite a few systems including Ag/GaAs, Ag/Si(111), and Pb/Si(111). The implication could be significant, since the formation of these uniform, self-organized atomic structures points to a potentially interesting pathway to prepare functional nanostructures. It is believed that this extra stability of metal films with specific thickness has an electronic origin, and can be explained by the so-called quantum size effects due to electron confinement. These quantum well (QW) states also give rise to an oscillatory work function as the thickness varies, and thus affect the details of the surface adsorption processes. In addition, the QW states are directly connected to the oscillation in the exchange coupling between two magnetic materials across a nonmagnetic spacer layer of various thicknesses. In this talk, I will present our recent density-functional calculations to study these effects in Ag/Fe(100), Pb/Si(111), and various freestanding films. The role played by the substrate and the crystal band structure will also be discussed.
*Supported by the National Science Foundation and the Department of Energy
Quantum confinement of electrons in nanostructures gives rise to most of their interesting physical properties. These are therefore important topics in materials simulations. In the following, two examples are given and the major findings are summarized.
An intriguing and unexpected feature has recently been discovered during epitaxial growth of metal thin films on semiconductors. Instead of forming three dimensional (3D) islands of various size as commonly observed for nonreactive interfaces, the metal atoms can arrange themselves into plateaus or islands of selective heights with flat tops and steep edges under certain growth conditions. This unusual behavior has been observed in quite a few systems including Ag/GaAs, Ag/Si(111), and Pb/Si(111). The implication could be significant, since the formation of these uniform, self organized atomic structures points to a potentially interesting pathway to prepare functional nanostructures. It is believed that this extra stability of metal films with specific thickness has an electronic origin, and can be explained by the so called quantum size effects due to electron confinement. As an example, the calculated relative surface energies for the freestanding and supported Pb(111) films exhibit an even odd oscillation as shown in Fig. 1.1 In particular, for lead films on a silicon substrate, the altered boundary condition produces a phase shift in the envelope function, resulting in deep minima at 1 monolayer and 6 monolayers. The absolute energy minimum at 1 monolayer explains the wetting layer, while the minimum at 6 monolayers accounts for the magic island height. For coverages between these two values, the system would be expected to phase separate into a linear combination of the two, in excellent agreement with the behavior observed in the experiment. These quantum well (QW) states also give rise to an os�cillatory work function as the thickness varies, and thus affect the details of the surface adsorption processes. The role of the substrate in confining these electrons is also studied.2
Another interesting system is the semiconductor nanowires. We investigate the structural, electronic, and optical properties of hydrogen passivated silicon nanowires along [110] and [111] directions with diameter d up to 4.2 nm from first principles.3 The size and orientation dependence of the band gap is investigated and the local density gap is corrected with the GW approximation. Quantum confinement becomes significant for d < 2.2 nm, where the dielectric function exhibits strong anisotropy and new low-energy absorption peaks start to appear in the imaginary part of the dielectric function for polarization along the wire axis (Fig. 2).
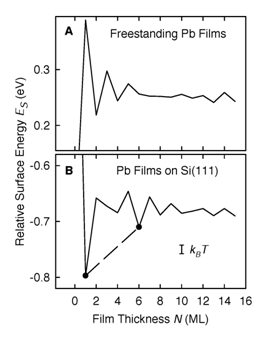
Figure 1. (A) Calculated relative surface energy per surface atom for freestanding Pb films as function of layer thickness, N. (B) The same for Pb flims on Si. In both cases, the energy reference is chosen in such a way that it is zero at N=0.
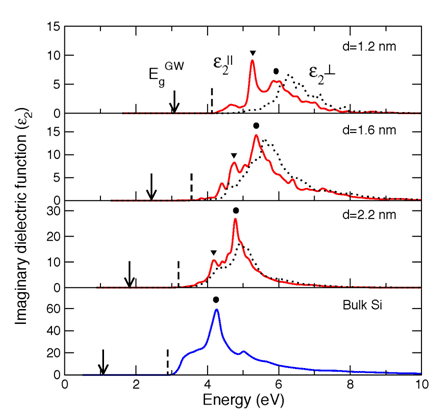
Figure 2. Imaginary part of the dielectric function, ε2(ω), polarized along the z direction (ε2||(ω)), solid lines) and in the xy plane (ε2⊥(ω), dotted lines) for silicon [110] wires with d=1.2, 1.6 and 2.2 nm, respectively. Shown in the bottom plane is ε2(ω) for bulk c-Si. The dielectric functions are calculated with the scissors operator to fix the band gap at the GW values (EgGW) and the energy zero is set to the top of the valence band. The arrows and vertical dashed lines mark EgGW and the optical absoprtion edges, respectively. The black dots indicate the original absorption peak in bulk Si, and the inverted triangles show the new peaks developed in nanowires.
Advances in modeling material systems since the development of quantum mechanics in the 1920's came much slower than progress in unraveling the electronic structure of atoms. This is particularly evident when one compares the identification of spectral features. For atomic spectra, lines are sharp and identification in terms of electronic transitions is much easier than for the case of solids where spectral features are generally broad. At first, empirical approaches paved the way, and eventually, it became possible to explain electronic and structural properties of fairly complex solids from first principles using only information about their constituent atoms as input. Because of the central role of electronic structure in understanding bonding and other properties, much of the focus has been on obtaining band structures and electron density maps. Eventually, this led to accurate determinations of ground-state mechanical and vibrational properties. In fact, at this time, ground-state calculations are of high precision and have been extended to compute electron-lattice interactions. In turn, these are used to explain and predict superconductivity in materials and to provide detailed calculations of superconducting properties. The model used for much of this work is based on pseudopotentials and density functional theory. It is sometimes referred to as the "Standard Model of Solids". The approach is a result of the development of many new conceptual models and the great progress in computation. Currently, excited states can be treated with excellent precision so that optical and photoemission data can be interpreted using first-principles theory. The fact that theory is now at a point where specific experiments for real materials can often be reproduced motivates a deeper and broader collaborative effort between experiment and theory. In addition, theory has produced successful predictions related to new materials and material properties. This has also enhanced experimental-theoretical collaborations. Some promising new avenues for research in the near term are the studies of the effects of confinement such as those arising in nanostructures, studies of systems where strong electron-electron correlation effects are dominant, exploration of multiscale properties going from nano to macro, etc. Much of the success of this research will depend on extending our current conceptual base and computational techniques. An example of the latter is the current effort to develop "order N" techniques to deal with more complex materials without overloading computers. The goals for researchers doing multiscale studies, molecular dynamics, and "order N" studies overlap, and progress is being made. Summarizing, there are a variety of opportunities in the field of modeling material systems ranging from development of new computational techniques to the invention of new concepts on how to view materials to explaining and predicting phenomena and properties. This presentation will survey the background in this area and explore some of the current proposals for future research.
The development of finite element simulation of material forming processes started about 30 years ago in academic laboratories, while the introduction of the corresponding commercial computer codes in industry is less than twenty years old. Numerical simulation is now a well-established tool for accelerating and improving design and optimization of material forming processes. It is currently used in industry for metals, polymers, glasses and other materials forming. From the computational point of view material forming is between fluid and solid mechanics and it combines both related techniques. This presentation will review the main achievements in simulation of complex process : bulk metal forming processes like forging using a Lagrangian approach with remeshing and injection molding using an Eulerian approach and VoF technique with extension to metal casting. Computational method and numerical techniques will be discussed: mixed finite element methods, Eulerian, convection diffusion solution, level set or VoF method, Lagrangian approach, remeshing and adaptive remeshing, anisotropic meshing. In the same time, the need of a more accurate modeling of the behavior of the material during flowing and also during its solidification requires to go further in the physical model contained in the constitutive equation. Moreover the future of material forming process simulation to go further in the prediction of the final properties of the work-piece, via the description of the microstructure evolution and using more often a multiscale modeling approach. Examples of complex multiscale material modeling will be discussed:
The magnetic properties of diluted magnetic semiconductors are calculated within the framework of the KKR-CPA, using a mapping on a Heisenberg model. Effective exchange coupling constants are evaluated by embedding two impurities in the CPA medium. Curie temperatures (Tc) are estimated by the mean-field approximation (MFA), the random phase approximation (RPA) and by Monte Carlo methods. In MFA and RPA, Tc is proportional to the square root of Mn concentration c for (Ga, Mn)N, while in (Ga, Mn)Sb Tc is linear to c. Since the extended hole states mediate the ferromagnetism in (Ga,Mn)Sb (p-d) exchange), the interaction is long range leading to a flat spin wave dispersion. Thus, the MFA gives similar results as the RPA. In (Ga, Mn)N, due to the broadening of the impurity bands in the gap, the ferromagnetic state is stabilized by double exchange. Since the impurity states are well localized, the exchange interaction is short range leading to pronounced percolation effects for smaller concentrations, which cannot be described by the MFA or RPA. Monte Carlo calculations show that the Curie temperature is strongly reduced as compared to the MFA values.

Calculated total spin moments for all studied Heusler alloys. The dashed line represents the Slater-Pauling behaviour. The blue circles denote ferromagnetic Heusler alloys which are not half-metallic and have a non-integer total moment, thus deviating from the S-P curve.
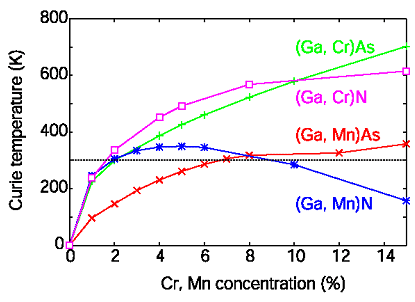
Calculated Curie temperatures of Cr- and Mn-doped GaAs and GaN. The dashed line indicates room temperature (300 K).
General Comments on the Relevance of Materials for Spintronics to the Workshop
The discovery of Giant Magnetoresistance (GMR) in 1988 initiated a tremendous worldwide series of discoveries: e.g. tunneling magnetoresistance (1995) in ferromagnet/insulator heterostructures, current induced switching (1999) and ferromagnetism of diluted magnetic semiconductors (1998). Commercial products based on GMR technology have been introduced in the market, and GMR read-out heads are now found in every computer. Non-volatile magnetic random access memories (MRAM) are the next important product. The vision is a superior spintronics, with higher speed, lower power consumption, excellent scalability and non-volatility.
The realization of this vision requires a completely new class of materials with large spin polarization at Ef, i.e., half metallic ferromagnets which have in one spin direction a gap at Ef, thus exhibiting a 100% spin polarization and raising the hope for extremely efficient spin dependent devices. Basically two classes of these systems exist: (i) Diluted magnetic semiconductors like Ga{1-x}Mn{x}As, which might lead to an all-semiconductor spintronics and (ii) half metallic alloys like Heusler alloys, manganites or double perovskites. Unfortunately these materials are extremely complicated, e.g. the diluted magnetic semiconductors are inherently instable and can only be produced by MBE. All these materials are multicomponent systems that contain many defects etc. Therefore ab initio calculations are urgently needed to assist the experimental research in this field. Such density functional calculations can provide a basic understanding of the electronic structure and can make valuable predictions for the magnetic and structural properties. Whole classes of materials can be screened in these calculations and hopeful candidates for spintronics applications can be identified.
For dilute magnetic semiconductors, the most important problem is a realistic determination of the Curie temperature. In experiments always clustering is observed and it is very different to distinguish the magnetism of the inclusions from the one of a coherent ferromagnetic phase. Although this is, due to the substitutional and orientational disorder, an extremely complicated problem, it can be solved by ab initio calculations, requiring, however, both a very accurate determination of the electronic structure, by e.g. including correlation effects, as well as a proper statistical treatment of the moment disorder. For half metallic alloys like Heusler alloys, zinc-blende compounds like CrAs or double perovskites, the most important aspect is the control of the impurity and interface states in the half-metallic band gap. Theses states strongly reduce the half-metallicity and one has to search for alloys and their interfaces to semiconductors, where such states are suppressed or do not occur at all. Here again, theory has the best chance to investigate broad classes of materials and to identify suitable candidates for applications.
In the field of electronic structure calculations Europe and the USA are by far the most important contenders, both as far as the quality as well as the quantity of work is concerned. Collaboration between both sides should therefore lead to important synergy effects. Many loose connections between interesting groups across the Atlantic exist, but real collaborations are due to funding problems difficult to realize. Often even invitations to workshops represent a big problem.
References on Spintronics Materials
In this talk, a brief overview of our recent work on modeling strength and plasticity of nanocrystalline metallic materials will be given, along with a cursory discussion of aspects relating to thermal stability of such materials. In a concluding part of the talk, modeling of severe plastic deformation, particularly by equal channel angular pressing, leading to extreme grain refinement will also be discussed. The models used are macroscopic in nature, but they import information obtained at dislocation dynamics scale and involve nanostructure-related parameters, thus providing a frame for linking various length scales.
General Comments on the Promising Areas to Address in US-EU Research Program
An interesting new aspect of materials design that is emerging and that should be reflected in the new program, even though it has not been discussed at the SF meeting is on the designing of materials with a special architecture, the constituents being not features of the microstructure, but rather specially shaped discrete bodies combined to a structure. In the case of the system we are working with, these elements are topologically interlocked, providing the material with very special properties, including enhanced resistance to fracture. There are several groups moving in this direction, such as Mike Ashby's at Cambridge, John Hutchinson's at Harvard, our group at Clausthal, etc. Development of a constitutive theory for such materials requires non-trivial models, e.g. non-standard Cosserat continua - something totally new and exciting. Alternatively, discrete or finite element techniques could be adjusted to describe the mechanical response of such materials. That would be an excellent playground for computational materials scientists. I believe it would be timely to include this aspect of CMS in the EU-NSF program.
Relevant References
Activities in the Area of Computational Materials Science
The main activities involve modeling and computer simulation of the mechanical behavior of bulk materials based on microstructure-related and physically motivated approaches.1 While the ultimate aim is to provide macroscopic continuum models suitable for FEM simulations, the design of a model is guided by elementary mechanisms of plasticity, notably at dislocation scale. The resulting models are simple in their architecture and very economical in terms of the number of parameters involved, yet robust enough to account for the macroscopic mechanical response. Another feature of the models developed so far is their modular structure, additional internal variables being introduced “on demand,” if needed to describe a new phenomenon of interest. An example is modeling of dynamic strain ageing, where the solute concentration on dislocations is introduced as an extra internal variable, in addition to the mobile and forest dislocation densities used in a basic, “skeleton” model.
Recent work concentrates on modeling of the mechanical behavior of nanostructured materials,2, 3 including aspects associated with grain growth and thermal stability of bulk nanomaterials.4, 5 The approach taken is based on treating grain boundaries as a separate “phase” and using a rule of mixtures approach, yet taking into account interaction between the two phases.
Significant progress has been achieved with modeling the evolution of microstructure, texture and strength of metallic materials subjected to equal channel angular pressing (ECAP). Again, a modified phase mixtures approach is used; the two “phases” considered being dislocation cell walls and cell interiors.6 Recent simulations of the ECAP processing are in very good agreement with the experimental findings (Fig. 1).7, 8
Another activity to be mentioned here is design of new material architectures based on topological interlocking of (identical) constituent elements. Such materials and/or structures (Figs. 2,3) possess a number of very promising features, such as unusually high tolerance to local fractures, sound absorption and, potentially, impact resistance.9, 10 FEM simulations of the mechanical response of these novel structures, along with efforts at developing appropriate non-standard continuum models are under way.
The Institute is linked to a number of groups dealing with development of new materials and with computational materials science in Germany, France, Australia, USA, Korea, Israel, Hungary and Russia and is open to further collaborative efforts in these areas.
Of particular interest to us would be to team up with groups doing molecular dynamics simulations of plasticity of nanomaterials, particularly with regard to interactions between dislocations and grain boundaries. A second area where collaborations are sought is evolution of dislocation cell structure under severe plastic deformation, including the development of misorientations and their role in strength of the material.
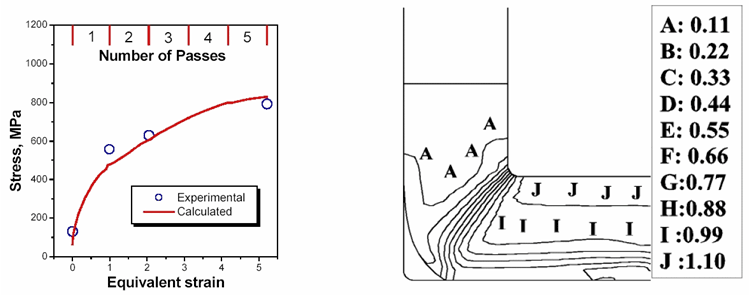
Figure 1. Strengthening of an IF steel by ECAP. Left: strength as a function of the number of ECAP passes (simulation vs. experiment); right: strain developing in a work piece during the first pressing.

Figure 2. Assembly of identical interlocked aluminum cubes showing anomalous mechanical response (negative stiffness upon unloading). Left: experiment, right: FEM simulation with ABAQUS.
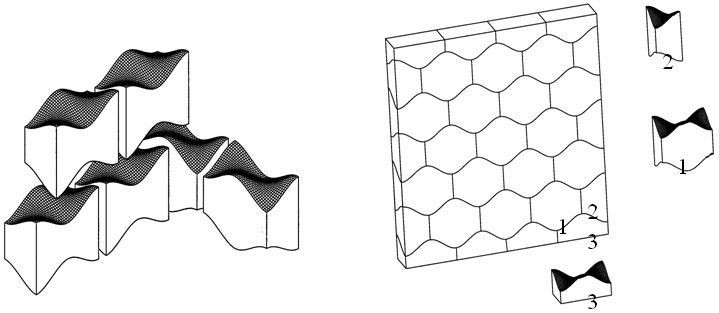
Figure 3. Assembly of a masonry structure. Left: principle of assembly of layer and corner structures; right: planar layer of osteomorphic blocks (1) completed with half-blocks (2), (3).
References
The evolution of the microstructure during the processing of the materials strongly influences their final properties. Thus, in addition to experimental data theoretical models are required to describe the changes in the microstructure. The theoretical models can be distinguished by their length-scale of resolution. At the continuum scale the sharp interface models are located. Both the precipitation kinetics of different carbides in steel and the austenite-to-ferrite phase transformation have been investigated by considering Onsager’s extremal principle of maximum Gibbs energy dissipation. The growth and shrinkage of carbides in a multicomponent steel matrix can be modeled qualitatively, details can be found in.1 A quantitative description of the precipitation sequences relies on experimental data, e.g. the surface energies of the precipitates, which are not yet available with sufficient accuracy. However, detailed experimental studies are in progress in order to compare the theoretical findings with the evolution of the phase fractions of the carbides. The austenite (γ)-to-ferrite (α) phase transformation in substitutional alloys has been modeled by assuming a sharp interface, too.2 Modeling approaches for substitutional and interstitial diffusion can be found in.3 Our model takes into consideration that the kinetics of the diffusional phase transformation depends on both energy dissipation by the bulk diffusion of the components and by a migrating interface. Onsager’s principle of maximum Gibbs energy dissipation is also applicable and, without any artificial constraints like local para- or orthoequilibrium, the evolution of the kinetic variables, the fluxes and the interface velocity can be calculated. In addition, it has been derived that the jumps of the chemical potentials of the components are all equal at a sharp interface. The situation in the substitutional alloy during austenite to ferrite phase transformation is depicted in Fig. 1. It is possible to simulate both mobility controlled as well as diffusion controlled transformation and the transient stages. An Fe-Cr-Ni alloy is considered with an initial composition of yCr = 0.004 and yNi = 0.02 (y denotes the site fractions), which transforms from austenite to ferrite at constant temperature of 1050K, see Fig. 2. This model [2] considers off-diagonal terms in the Lik-matrix, where Lik are the Onsager coefficients directly related to the tracer diffusivities of the components. Substitutional diffusion is assumed to occur by a change of the lattice sites of neighboring atoms in this model, vacancies are not taken into account yet. In order to implement the more realistic vacancy mediated diffusion into this model; the quasi-tracer diffusivity A0 of vacancies has to be determined. In a recent work of Hartmann et al.4 the coefficient A0 is determined by Monte-Carlo-simulations. Constant atomic fluxes are imposed and the gradients of the site fractions are measured. As far as the gradients and the fluxes of the components are linearly combined by the Onsager coefficients, the quasi tracer diffusivity of vacancies A0 can be obtained by this novel method.

Fig. 1: Schematic representation of the γ / α phase transformation in e.g. Fe-Cr-Ni alloys. The jumps Δ of the chemical potentials across the interface are equal for all substitutional components.
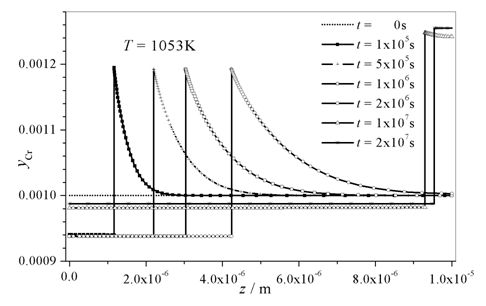
Fig. 2: Site fraction profiles of Cr at different transformation times. Isothermal thickening of ferrite plates in an Fe-Cr-Ni alloy (yCr = 0.004 and yNi = 0.02) at 1050K is simulated, the initial austenite grain size is 20m.
Views on US-EU Collaborations
It is of great value for our group to work together with scientists from USA to compare the simulations and results with each other and learn more about complex processes in the field of non-equilibrium thermodynamics.
References
The role of computation in understanding experimentally observed behavior of submicron semiconductor material structures will be illustrated through brief descriptions of several examples drawn from recent work. The examples include aspects of behavior that have been observed in either fabrication or functional performance of small structures and that arise through the influence of mechanical strain on essential characteristics of the material system. The examples include (i) the origin of the (105) crystallographic surface orientation that dominates morphology in the early stages of formation of SiGe/Si(100) quantum dots by strain driven self assembly, (ii) the evolution of arrays of islands of these same materials during vapor deposition, including island interactions, and (iii) the modification of current-voltage transport characteristics of a resonant tunnel diode due to inhomogeneous strain distributions induced through fabrication. The computational approaches used in these studies include molecular dynamics or first principles analysis of the structure of strained crystal surfaces in example (i), the incorporation of anisotropic surface energy into a continuum model of morphology evolution in example (ii), and illustration of the use of deformation potential theory within the effective mass model of quantum transport in example (iii). In each case, the synergy among experiment, theory and computation has been crucial to resolution of issues of importance for the advancement of certain nano electronic technologies.
Strain and surface effects in material nano structures
The study of material nanostructures at a basic level offers the promise of major advances in the understanding of electronic, optical and/or mechanical properties of materials, as well as for the manufacture of electronic or optical devices with unprecedented performance characteristics. From the fundamental point of view, the experimental determination of transport characteristics of small devices provides a spectroscopic window on their electronic structure. From the point of view of applications, on the other hand, insertion of these minute structures into practical devices could result in a dramatic increase in feature density, a decrease in power consumption and a reduction in response time. To realize these scientific and engineering benefits, it will be necessary to overcome some major challenges through broad and interactive programs of research. Success in this endeavor hinges on adopting a new paradigm concerning the interrelationships among material characteristics, fabrication methods and functional performance. The details of the conceptual framework will necessarily be provided through synergy between experiment, modeling and computation.
Strain-driven nucleation, growth and coarsening in epitaxial islands on a single crystal substrate holds promise as a means of manufacturing nanoscale devices. This approach is already in use to produce optical detectors consisting of arrays of quantum dots, with the detector bandwidth being controlled by the distribution of island sizes incorporated in the array. The use of isolated island quantum dots, embedded within an appropriate matrix material, as transistors and diodes has been demonstrated, but the technology for accessing or gating individual islands within arrays of dots remains elusive. In any case, progress in this area of research, particularly on the SiGe/Si material systems, provides a good illustration of the productive synergy among theory, experiment and computation that underlies progress.
When islands were first identified in the early stages of film growth by vapor deposition, theories based on diffusive morphology evolution were introduced to describe surface stability and shape change (Freund and Suresh 2003). As observations of islands became more refined, it was discovered that the lateral faces of the hut shaped islands in the early stages of growth (up to impingement of islands on each other, say) were invariably (105) crystallographic surfaces (Floro et al. 1997), and they arose only when the mismatch strain was compressive (Xie et al. 1994), both features being at odds with models of surface evolution prevailing at the time.
To resolve the discrepancy, detailed calculation of the formation energies of several surface step structures on biaxially strained Si and Ge (001) surfaces were computed by both first principles methods and molecular statics (Shenoy et al. 2002). Through this computational approach, it was discovered that a novel rebonded surface step, with a (100) orientation and with height equal to a quarter the unit cell dimension, is strongly stabilized by compressive strain compared to all other surface reconstructions which have been proposed previously. Furthermore, the minimum energy orientation of vicinal surfaces composed of such steps was found to be a (105) crystallographic orientation. It could be concluded that the lateral faces of the hut shaped islands observed in experiments on SiGe/Si (100) faces are composed of these steps. In addition, the calculations provided an explanation for the absence of any kind of nucleation barrier for the formation of islands, as had also been recently established experimentally (Sutter and Lagally 2000). Subsequently, a variational approach as a basis for studying the nonlinear dynamics of nanoscale surface undulation was developed as a basis for large-scale numerical simulations. In this way, the dependence of surface energy on strain and surface orientation can be completely determined by the features of atomistic simulation of discrete material structures and incorporated into larger scale simulations, without the need for arbitrary ad hoc assumptions.
The experience gained with this particular material system has opened the way for the study of other phenomena by the same general approach of integrating experimental observations and careful material simulation studies to understand physical phenomena of interest. Examples of potentially fruitful problem areas include:
Drucker, J. (2002), “Self-assembling Ge(Si)/Si(100) quantum dots,” IEEE Journal of Quantum Electronics 38, 975-987.
Floro, J. A., Chason, E., Lee, S. R., Twesten, R. D., Hwang, R. Q. and Freund, L. B. (1997), “Real-time stress evolution during Si{1-x}Ge{x} heteroepiaxy: Dislocations, islanding and segregation,” Journal of Electronic Materials 26, 969-979.
Freund, L. B. and Suresh, S. (2003), “Thin Film Materials,” Cambridge University Press.
Fujikawa, Y., Akiyama, K., Nagao, T., Sakurai, T., Lagally, M. G., Hashimoto, T., Morikawa, Y. and Terakura, K. (2002), “Origin of the stability of Ge (105) on Si: A new structure model and surface strain relaxation,” Physical Review Letters 88, 176101.
Kamins, T. I., Li, X. and Williams, R. S. (2004), “Growth and structure of chemically vapor deposited Ge nanowires on Si substrates,” Nano Letters 4, 503-506.
Ma, D. D. D., Lee, C. S., Au, F. C. K., Tong, S. Y. and Lee, S. T. (2003), “Small-diameter silicon nanowire surfaces,” Science 299, 1874-1877.
Shenoy, V. B., Ciobanu, C. and Freund, L. B. (2002), “Strain induced stabilization of stepped Si and Ge surfaces near (001),“ Applied Physics Letters 81, 364-366.
Sutter, P. and Lagally, M. G. (2000), “Nucleationless three-dimensional island formation in low misfit heteroepitaxy,” Physical Review Letters 84, 4637-4640.
Vandervelde, T. E., Kumar, P., Kobayashi, T., Gray, J. L., Pernell, T., Floro, J. A., Hull, R. and Bean, J. C. (2003), “Growth of quantum fortress structures in Si {1-x} Ge {x}/Si via combinatorial deposition,” Applied Physics Letters 83, 5205-5207.
Wu, Y., Cui, Y., Huyanh, L., Barrelet, C. J., Bell, D. C. and Lieber, C. M. (2004), “Controlled growth of structures of molecular scale silicon nanowires,” Nano Letters 4,433–436.
Xie, Y. H., Gilmer, G. H., Roland, C., Buratto, S. K., Chang, J. Y. and Fitzgerald, E. A., Kortan, A. R., Schuppler, S., Marcus, M. A. and Citrin, P. H. (1994), “Semiconductor surface roughness: Dependence on sign and magnitude of bulk strain,” Physical Review Letters 73, 3006–3009.
Fabrication of next generation materials and devices comprised of molecular and nanoscopic building blocks tailor-made for specific applications will rely to a great extent on processes of self-assembly, in which instructions for organization emerge from the nature of the forces acting between constituents. Ideally, self-assembly will lead to synthetic structures whose form and function are explicitly encoded in the system at the molecular level, as in biological systems. However, the intermolecular forces required to produce organized assemblies of synthetic nanoparticles and nanostructured molecules with the precision and reliability typical of biological structures are not understood. Developing new approaches to self-assembly, especially bio-inspired approaches, is one of the most fundamental challenges of nanotechnology. Computational materials science has much to contribute to this exciting arena through the development of suitable theories, models and simulation methods capable of spanning from the scale of individual atoms to mesoscopic assemblies. Through simulation, it is possible to elucidate the fundamental principles of self-assembly and the properties of self-organized structures. In this talk, we present our recent molecular and mesoscale simulation studies of the selfassembly of collections of anisometric, amphiphilic nanocrystals and nanostructured molecules, and discuss opportunities for progress and the challenges inherent to these and other problems in computational nanoscience.
A phase field theory of polycrystalline solidification is presented that incorporates noise-induced nucleation of grains with different crystallographic orientations, the effect of foreign particles, and polycrystalline branching with fixed orientational misfit. Our model builds on previous work for nucleation and polycrystalline growth in phase field theory.1, 2 It will be shown that increasing the number of particulate additives and reducing the rotational diffusion leads to similar morphological transitions between single crystal dendrites and polycrystalline seaweed patterns. The formation of other complex polycrystalline morphologies (dizzy dendrites, spherulites, sheaves, axialites), observed in highly undercooled melts, will also be addressed (see Figure).
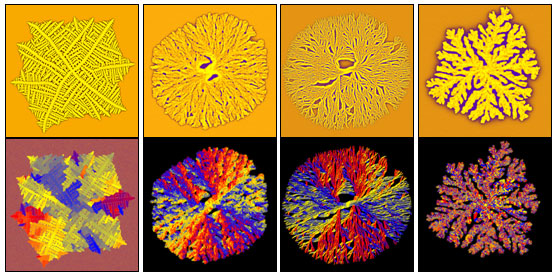
Polycrystalline solidification morphologies as predicted by the phase field theory. From left to right: A “dizzy” dendrite caused by particulate additives,2 two spherulites obtained with 30 and 15 degree branching, and a fractal-like aggregate. Chemical composition (upper row) and orientation maps (lower row) are shown. (Coloring: upper row: solidus — yellow, liquidus — blue; lower row: different colors correspond to different crystallographic orientations.)
References
Modeling and simulation of micro-casting processes with commercial tools is limited to simple materials. When it comes to process feedstocks from ceramic nanopowders in cavities with dimensions of a few tens of micrometers or even below the important geometrical features in the mould perilously approach the size of the solid particles in the material. On the other hand the injection moulding machine is a macroscopic object and has macroscopic material reservoirs. To tackle the flow of multicomponent materials all the way down from macroscopic reservoirs to the last microscopic cavity needs to account for various material properties. The simulation process with a continuum model will not perform well on the micro-scale, as well as microscopic particle based methods will be prohibitive in their application to macroscopic geometries. Therefore we propose a combination of particle based methods and continuum models for the simulation of this scale spanning problem. In an outlook we will indicate the usefulness of our approach to similar challenges.

This presentation surveys recent progress and experience with space-time discontinuous Galerkin (SDG) models, emphasizing their use as a practical tool and a conceptual framework for multiscale materials modeling. Discontinuous Galerkin finite element models are constructed with basis functions that are discontinuous across element boundaries; the Galerkin projection enforces weakly the jump conditions generated by the continuum balance laws and by kinematical compatibility. In the space-time version, an unstructured mesh partitions the analysis domain in†Ed � R, and the relevant balance laws and jump conditions are enforced directly on each cell and across each cell boundary. When applied to hyperbolic problems, SDG methods satisfy the balance laws to within machine precision on every element; they feature linear complexity in the number of elements, superior stability properties, and a rich parallel structure. Thus, they can provide high-resolution approximations in demanding materials-related applications. Space-time discontinuous Galerkin methods are an attractive option for multiscale continuum simulations. They facilitate hp-adaptive finite element methods that bridge multiple length scales, because they admit nonconforming grids and nonconforming basis functions in adjacent elements. When applied to sharp interface models for material phase boundaries, SDG meshes can continuously track moving interfaces without suffering the numerical errors associated with element distortion and data projection that plague conventional moving grid methods. Their superior shock-capturing capabilities make SDG models an attractive choice for dynamic simulation problems, such as Austenite-Martensite transitions in shape memory alloys subjected to shock loading (whether by a sharp or diffuse interface model) and in certain forms of dynamic fracture. Cohesive fracture models are easy to implement, because displacement discontinuities across element interfaces are part of the basic SDG framework. We discuss prospects for the use of SDG methodologies in atomistic modeling and as a bridging mechanism in simulations that attempt to couple atomistic and continuummodels. For example, our parallel and spacetime-adaptive†O N ( ) solution technology can be used to solve the Schr�dinger equation in time-dependent density functional theory. Beyond discontinuities in the continuum fields, the SDG framework can be used to address discontinuities in the physical model itself. In contrast to purely kinematical coupling, we are investigating weak SDG formulations that couple continuum and atomistic models by enforcing weakly the jump conditions from physical balance laws.
Growth is a classic example of a multiscale problem. At the continuum scale, there are problems of lattice matching and the generation of stress fields. At the atomic scale, there are issues of the detailed mechanisms involved in the growth process. The problem of reaching the timescales required is particularly serious when attempts are made to simulate growth. Molecular dynamics simulations of MBE often assume growth rates of 104-106 times those available to any experimentalist. Attempts to simulate nucleation events can run into even more drastic problems. We consider two examples, one from hard material interface, one from a hard/soft interface to illustrate the problems. A simple example of the growth of one material on another is the growth of thin layers of ceramic oxide on a ceramic substrate. The difficulty here is that the barriers for processes are often high. We require a method that can reach the timescales needed without assuming what the processes involved are. We have demonstrated that a combination of the temperature-assisted hyperdynamics scheme of Voter and coworkers2 with kinetic Monte Carlo achieve this. The simulations show the importance of a wide range of cooperative transport mechanisms that have usually been ignored in ionic diffusion. Our simulations show that exchange mechanisms can dominate diffusion of ionic molecules over the surfaces of oxides with the rock salt structure. We also discuss the important issue of whether it is possible to grow atomically sharp interfaces in ceramic hetero-interfaces, or whether the exchange mechanism makes mixing unavoidable. An example that combines the problems of interfacial lattice-matching and nucleation is that of biomineralization. Living organisms can control the size, shape and structure of minerals. Attempts to reproduce this biological control in the laboratory often use Langmuir monolayers of long-chain carboxylic acids.3 It is usually assumed that the organic layer acting as a template, which controls the growth morphology of the mineral, exercises this control. We use a combination of large-scale molecular dynamics simulations and the Wulff-Kaishew theorem to predict the morphologies of calcite crystals grown on stearic (octadecanoic) acid monolayers and find good agreement with experiments. We show that, while templating ideas are important, it must be remembered that organic monolayers are not rigid structures in a vacuum. They are flexible, chemically active surfaces in contact with water. Any theory of mineral growth must include these effects. A case of particular interest is when a mineral grows in a polar direction. This introduces a new constraint (the necessity to quench the macroscopic dipole) that must also be included.
Relevant References
Interface processes and film growth in ceramics
Ultra-thin films can display structures and properties very different from bulk materials of the same composition. The constraints imposed by the substrate can stabilize new and different structures. The control that MBE offers means that such structures can be designed. Films with a macroscopic dipole (polar directions) can be grown because they have finite thickness, and so the energetic considerations that prevent their appearance in the bulk do not apply. The resulting control of the potential suggests that band offsets can also be controlled. Where structural phase transitions occur, transition temperatures often depend on the thickness of the epitaxial layer. Computer simulation has a vital role to play in interpreting the experimental data and in predicting structures that can arise.
There are serious limitations to current simulation techniques. Lattice energy minimization permits the study of possible structures, but not the dynamics. Kinetic Monte Carlo can reach the relevant timescales, but traditional implementations require a list of possible processes. Both these approaches risk bias on the part of the simulator. Direct MD simulations remove bias, but at the cost of using unrealistic timescales (~1010 monolayers/sec compared with experimental deposition rates of <1 monolayer/sec). Available computer power is still rapidly increasing, but there is no possibility of a direct simulation of film growth at a realistic rate. When transition-rate theory is applicable, the hyperdynamics methods introduced by Voter and co-workers show great promise for these problems. The most practical and robust method currently is temperature-accelerated dynamics (TAD). Ceramics are ideal candidates for this method; the barriers to ion motion are high, harmonic transition state theory is a good approximation and (useful for TAD) the system can often be run at high temperature. The alternative bias-potential methods have serious problems. The “puddle-skimming” methods have a simple potential, but it is difficult to position it correctly. More complex potentials have numerical instabilities. A good solution to the problem of defining the bias potential would make this the hyperdynamics technique of choice.
Informal US/EU collaborations between Voter (Los Alamos) and European groups already exist. A recent CECAM workshop on accelerating dynamical simulations (organized by Allen (Warwick) and Kneller (Orleans)) brought together many EU groups interested in this topic and highlighted current problems. (I can provide a list of participants and titles of talks if requested). A major US/EU effort in this area is therefore very timely. A particular advantage for long time-scale dynamics is that it could combine EU (particularly UK) expertise in the simulation of ceramic systems with US expertise in accelerated timescale methods.
Structural modeling of the biological interface with materials
Modeling bio-material interfaces and biomaterials is a major problem requiring collaboration between groups that are world-class in both simulation and experiment. It has immediate impact on medicine, biomimetics and nanotechnology and is likely to have spin-off benefits for the oil and pharmaceutical industries. Bio-material interfaces often determine biomaterial properties. Molecular recognition of surfaces by proteins is critical for biocompatibility since the attachment of cells to implants is initiated by protein adsorption. The development of biosensors and the full exploitation of probe microscopies for chemical resolution need detailed models of bio-material interfaces. Furthermore, the control of biomineralization on organic scaffolds and templates is a key to the directed self-assembly that is essential if devices based on molecular-scale electronics are to be mass-produced. While recent results demonstrate clearly that proteins can control the assembly of nanoparticles, further progress requires an understanding of the specifics of interface recognition.
Modeling the energetics and kinetics of bio-material interfaces in realistic aqueous environments is an essential step towards this understanding. This requires the solution of two problems at different scales. At the atomistic scale, the problem is the lack of accurate force-fields, while simulation of nucleation and growth at an interface requires new developments in mesoscale modeling. Simulating such systems is a multi-disciplinary problem, requiring expertise in materials such as ceramics, biomolecules and polymers as well as techniques from ab initio simulations to classical molecular dynamics, long time-scale dynamics and mesoscale modeling.
There are three fundamental scientific challenges: developing new force-fields and protocols, dynamic models of the bio-material interface and new models of nucleation and self-assembly. New force-fields are particularly needed for the organic/inorganic interface. The usual combining rules for organic molecules do not work well (particularly for interface binding energies). There are therefore no reliable protocols for generating force-fields for a given system. The two further challenges have the common problem of long times-scales. Direct simulations of spontaneous nucleation processes like crystallization show the limitations of classical nucleation theory. Yet such simulations are limited to timescales many orders of magnitude shorter than the induction time of crystal growth processes. Kinetic Monte Carlo methods avoid this problem but do not solve it since they assume a mechanism for crystal nucleation and growth. Many computational schemes use the idea of coarse-graining to the mesoscale. This involves a major problem in mapping from one scale to the next. Nucleation is a particular problem since formation of a critical nucleus may involve a multi-dimensional reaction coordinate that is difficult to predict a priori, and involves molecular detail that is obscured in mesoscale methods. New methods are needed to treat the soft, reorientation and translational motions relevant here.
The problems of simulating biominerals and biomineralization involve generic issues that are being tackled by many groups and groupings on both sides of the Atlantic. There are major experimental groups both in the EU and the USA and large amounts of experimental data now available that need interpretation. A US/EU modeling collaboration in this area could make a major impact in the field.
References
Figure 1. Exchange mechanism for diffusion of an ionic molecule on the (100) surface of BaO (top and side views). Red and grey spheres represent the oxide and barium ions in the (100) surface respectively. Green and blue spheres denote the oxide and barium ions in the BaO molecule. The yellow sphere is the barium ion displaced by the exchange mechanism.
Figure 2: Evolution of island growth on the surface of BaO (Kinetic Monte Carlo simulations)

Figure 3: Structure of small calcite cluster (012 orientation) growing on a self-assembled monolayer in water.
This talk will concentrate on numerical modeling of fracture problems in polymers. Finite Volume (FV) method will be briefly reviewed particularly its application to coupled solid-fluid-fracture problems, and the main topic of the talk will be on the issues related to the accurate prediction of fractures independent of the numerical method used. Particular emphasis will be paid to: a) Prediction of fracture in ductile PE, and b) Prediction of fast cracks and crack instabilities in brittle PMMA. Cohesive Zone Model (CZM) was employed in both cases to describe the failure criteria in terms of a traction-separation law. In the former case, attempts were made to experimentally determine CZM properties as a function of rate and constraint. In the latter case where such measurements were not possible due to small time and size scales, the traction-separation law was numerically calibrated based on experimental observations. It is demonstrated that precise knowledge of CZM parameters is of crucial importance in predicting the fractures. It was argued that use of Molecular Dynamics may be very useful in understanding the mechanisms of crazing, which is believed to be the main mechanism of damage leading to fracture, and therefore to quantify CZM parameters. Structural properties such as molecular weight, entanglements, branching etc could then be explicitly accounted for.
Views on US-EU Collaboration: Modeling of Fractures in Polymers and Adhesives Following the EU-USA workshop in San Francisco on 15-16 April ‘04 on Computational Material Modeling, I would like to express some views on the necessity and urgency for collaborative EU-USA work in the area of fracture in polymers and adhesives. First of all I must say that I found the meeting extremely useful and interesting. It became obvious that, although the resources are available and benefits are enormous and clear, there is no much interaction and information exchange between research groups working on various scales from molecular to continuum. I had long discussions with two leading experts in Molecular Dynamics (MD) simulations of fracture and crazing in polymers and we agree that the best way forward in the area of deformation/damage/instability/failure of polymers and adhesives is by linking MD with continuum approaches. Particular emphasis was on predicting the failures via embedded Cohesive Zone Models, which represent the complete evolution of various damage mechanisms leading to final failure in a simple collective manner via traction-separation laws. The effects of “global” parameters such as constraint, rate and temperature are particularly important in polymers, as well as the effect of molecular structure, i.e. molecular weight, entanglements, branching, cross-linking, etc. There is a vast amount of work where CZMs are employed in predicting failures of polymers and adhesives (as indeed metals and other structural and functional materials), and vast majority uses assumed CZM parameters without having much insight into their physical characteristics. Only in few cases attempts have been made to characterize the CZM parameters experimentally or from micro-mechanical models. I propose to establish the collaboration with MD, micro and macro approach groups to study the evolution of damage (crazing in particular) in polymers (PE and PMMA) as well as in epoxy-based rubber modified adhesives. This collaboration will aim to achieve one (or more) of the following:
We already started serious talks on identifying six or more EU-USA collaborators.
Some relevant own papers and references from potential collaborators:
This presentation will focus on crack propagation in glasses, nanostructured ceramics, and nanocomposites. We have performed multimillion-atom molecular dynamics simulations to investigate the morphology and dynamics of crack fronts in these systems. The results on atomistic mechanism of fracture in glasses are in excellent agreement with recent AFM studies. Roughness exponents of fracture surfaces are also determined and the results are in accordance with experiments.
Computational Materials Science focuses on understanding the magnetic, optical, electrical and mechanical properties of materials and the relationship between their physical properties, chemical composition and atomic structures. It is based on ab initio electronic structure calculations that use only our knowledge of quantum mechanics and the fundamental physical constants to interpret experiments using a minimum of experimental input, and to make material-specific predictions. Three main themes can be identified: ground state properties, electronic transport, and electronic excitations. In this presentation, I outline the current state of the art in regard to parameter-free electronic transport calculations.
References
Ground State Properties
Transport
Excitations
Complex fluids such as amphiphilic mixtures, colloidal suspensions, and polymer solutions, mixtures, and melts are characterized by structure on mesoscopic length-scales: ranging from nano- to micrometers and energy scales comparable to the thermal energy kBT. The meso-scale structures of these systems endows them with many interesting and unique features, and they are widely used in the processing, chemical, and energy industries. Complex fluids present a challenge for conventional methods of simulation due to the presence of disparate time scales in their dynamics. The unique problems associated with the modeling and analysis of the behavior of these systems has created the need for new simulation techniques that overcome some of the difficulties associated with the use of atomistic molecular dynamics simulations and macroscopic approaches based on the numerical solution of continuum equations. The modeling of these systems requires the use of "coarse-grained" or mesoscopic approaches that mimic the behavior of atomistic systems on the length scales of interest. The goal is to incorporate the essential features of the microscopic and mesoscopic physics in models that are computationally efficient and are easily implemented in complex geometries and on parallel computers. Recent research involving the development and application of a range of mesoscale simulation techniques will be summarized. In particular, work involving the use of coarse grained dynamically triangulated surface models of membranes to analyze flicker spectroscopy data of giant nonspherical vesicles, the budding of crystalline (clathrin-coated pits) in fluid membranes, and the phase behavior and structure of microemulsions. Membranes of fluctuating topology will be described. A range of applications of recently developed particle-based mesoscopic simulation techniques will also be discussed. Some future research involving extensions of these techniques to model the dynamics and rheology of complex mixtures is outlined.
Basic properties in relatively simple materials are controlled by a single process taking place in highly random or ordered structures (e.g. elasticity of ordered intermetallics, plastic deformation of micro-grained polycrystals). Modern materials have increasingly complex structures at different length scales and are designed for multi-functional applications. Their properties are controlled by structural elements which form stochastic populations. Models are needed which take into account stochastic (variable) features of relevant structural elements and their interactions. Nano-materials are one of rapidly growing fields of materials research. Size of the elements by definition in this case is of prime importance. Despite that the development in this field is currently mainly of experimental nature. The quantitative description of the size, shape and arrangement of nano-elements receives frequently marginal attention. In order to assure expected progress in the development of nano-materials, models are needed, which could be used to explain properties of materials containing varying fractions of nano-objects (grains, particles) differing in the size, shape and arrangement. In particular size effect in the range of nano-metric dimensions should be explained for systems containing stochastic populations of grains and/or particles. Modeling of nano-systems of technical importance requires multi-length scale and stochastic approach. Complexity of the structures ranging from clusters of atoms up to aggregates of nano-particles and grains calls for internationally coordinated efforts. In particular, modeling of size effect in stochastic assemblies of nano-particles and grains is suggested as a starting point for US-EU collaboration.


(a) Modeling based on representative (codified) elements and effects of (b) homogeneous and (c) inhomogeneous grain size distributions in (d) grain growth kinetics
Calculation of the various properties of materials often requires very different theoretical and computational approaches because of the complex interactions and diverse behaviors in condensed matter. In particular, the restricted geometry and symmetry of nanostructures often give rise to interesting quantum confinement, enhanced many-electron interaction, and other effects related to reduced dimensionality. These effects can lead to novel physical properties and phenomena, which also are potentially useful in applications. In this talk, I will discuss some of our recent studies on the electron transport, optical, and mechanical properties of nanotubes and molecular junctions. For examples, molecular electronic devices, i.e., electrical transport through a single molecule can produce highly nonlinear I-V characteristics. The optical spectra of small diameter carbon nanotubes exhibit dramatic excitonic effects. Also friction on the nanoscale may be very different from that on the human scale. The talk will be on the theory and computation of these phenomena. We will present results on the nonlinear transport behavior of molecular junctions calculated using a newly developed ab initio scattering-state method, the optical response of nanotubes employing a first-principles many-particle Green's function approach, and the behaviors of mechanical energy dissipation in double-walled carbon nanotube oscillators from molecular dynamics simulations. The physical origin of the calculated behaviors will be examined.
The fracture of macroscopic brittle materials is sensitive to details at the atomic scale. I will discuss two implications of this idea. First, I will discuss ideal brittle twodimensional materials as a testing ground for the understanding of fracture, and show that solvable models violate established rules for how cracks move. Second, I will discuss efforts to bring theory and experiment into accord for the fracture of single-crystal silicon, and show that theory and experiment remain disconcertingly far apart.
Problems in Computational Materials Science
The continual growth of computers means that the number of degrees of freedom that can be described in computational materials problems increases at a rate given by Moore.s Law. However, the general experience of solving a computational materials problem has not changed as much as one might think, or hope, in the last ten years. The time needed to solve problems is dominated by writing code in C or Fortran, perhaps with an added layer of complexity introduced by MPI, or with an object-oriented wrapper in something like Python. In contrast to fields like numerical analysis, there is little community code or infrastructure. For density functional theory, there are some commercial codes, such as VASP, that seem to be widely used, mainly as black boxes. Moving up one phenomenological level to molecular dynamics, there is little software suitable for materials problems either commercially or publicly available. A small number of groups have codes that they use and develop internally. Many are willing to share code (CCP5 has molecular dynamics, but not parallel and without three body interactions, Sethna and others at Cornell have a Digital Materials project, I have posted a parallel molecular dynamics code with three body interactions at hola.sourceforge.net), but the lack of proper documentation, and friendly procedures for modifying and generalizing code seem to make it easier to start continually from scratch than for one group really to make use of what another has done.
Perhaps because various groups are continually building infrastructure from scratch, there are some deeper problems that seem continually to be left unaddressed. I would summarize them in the following way: The validity of the approximations underlying computations claiming to result from first principles is too rarely clear, and too rarely checked against experiment. I believe that this statement applies to density functional theory in the sense that density functional codes contain no internal flags warning the user that correlation effects are rendering the results delivered by the code especially inaccurate. Using the codes requires physical insight and expertise, making the availability of black box codes worrisome, since writing one is probably the best way to become aware of all the hidden pitfalls. I am sure it applies to molecular dynamics codes, which are the sort I know the best. The basic problem with Molecular Dynamics is that no one is quite sure just how much of reality it can represent in principle, let alone how much existing codes capture in reality. Are there quantum mechanical effects among interacting atoms that are significant for macroscopic mechanical behavior and that no classical force laws will ever get right? Not clear. Maybe some classical rules can be cooked up that will deal effectively with phenomena that seem intrinsically quantum mechanical, such as light emission from solids when the BornOppenheimer approximation fails, or maybe not.
I have been working on the problem of fracture of brittle materials for some time on the grounds that it should serve as a test case where one can establish what is understood and what is not. The simplest test case should be the fracture of single crystal silicon along its natural fracture planes at low temperature. Even for this simplest of all fracture problems, the discrepancy between theory and experiment remains discouragingly large. An early survey of experimental and theoretical results for initiation of cracks in silicon was carried out by Spence, Huang, and Sankey;9 experiments and theoretical predictions for dynamic fracture from me and my coworkers are in.4, 5, 6 Several other groups have studied the silicon problem as well.8, 3, 2, 7, 1, 10 Sometimes groups report the results of dynamic fracture studies by dividing the energy needed for a crack to propagate by the energy needed for it to initiate. Making use of this scaling parameter brings theory and experiment into fairly close accord. However, if one does not avail himself of this rescaling, then a summary of the current comparison of theory and experiment is as shown in Figure 1. The work of Perez and Gumbsch provides hints that quantum mechanical calculations simply give different results from all existing classical computations near the crack tip; however, the matter has not been pursued far enough to be resolved.
If resolving difficulties in this sort of idealized test case is hard, then much more work remains before it will be possible to give the reliable answers to questions about complex nonequilibrium materials that people outside the computational materials community expect us to deliver.
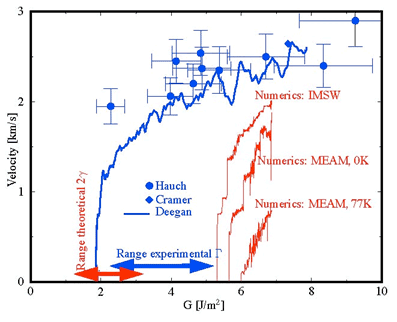
Figure 1: Comparison of theory and experiment for dynamic fracture of single crystal silicon. Theory and experiment differ by around a factor of two in predicting the energy for cracks to move at various speeds. According to almost all calculations, the energy needed for a crack to move dynamically (red lines) is around double the thermodynamic minimum energy (red arrow) needed in principle for cracks to begin moving. In experiment, cracks appear to begin moving dynamically (blue lines) almost exactly when at the thermodynamic lower bound (blue arrow).
References
Much of the current progress in many fields of materials science is owed to insights obtained from recent large-scale atomistic simulations. However, these cannot be evaluated in isolation with respect to conventional engineering approaches (FEM, elasticity) and experimental observations (TEM, PAS, etc). In this talk, two aspects related to dislocation loop embrittlement and dislocation glide in Fe are discussed with emphasis placed on bridging experiments with multiscale modeling. The first part deals with the nucleation and growth of �100� interstitial loops in α-Fe. Although widely observed experimentally, �100� loops are known to be higher in energy as calculated from Frank's rule and MD observations, and, thus, their existence has remained a puzzle for decades. Here a comprehensive mechanism is presented that reconciles the current understanding on loop formation in Fe from the standpoint of atomistic simulations, continuum elasticity and experimental observations. Qualitative comparison among experimental micrographs and atomistic structures is provided by way of a TEM image simulation method. The second part is concerned with screw dislocation motion in Fe. The first observations of the long-hypothesized kink-pair mechanism in action are obtained in atomistic simulations of dislocation motion in Fe. However, in a striking deviation from the classical picture, dislocation motion under higher stress becomes rough resulting in spontaneous self-pinning and production of large quantities of debris. Then, under still higher stress, the dislocation stops abruptly and emits a twin plate that immediately takes over as the dominant mode of plastic deformation. These observations challenge the applicability of the Peierls threshold concept to 3-dimensional motion of screw dislocations and suggest a new interpretation of plastic strength and microstructure of shocked metals.
I shall briefly some application of coarse-grained models to explore the properties of interfaces and surfaces of soft matter, e.g., the stability of thin polymer coatings,1, 2 wetting properties3 and self-assembly in multi-component polymer films, and the properties of bilayer membranes.4 We investigate these systems by Monte Carlo / Molecular Dynamics simulations and by self-consistent field theory / density functional theory for polymeric systems. Both, the structure and thermodynamics as well as dynamical properties in thin films have been studied by our coarse-grained models.
References
Stability and structure formation in thin polymer films
Interfaces and surfaces play an important role in soft matter: For instance, controlling the free energy of surfaces and interfaces,1–3 one can prevent polymer coatings from dewetting. The morphology of a phase-separated polymer blend can be conceived as an assembly of interfaces, adding bock copolymers4 one can fabricate disordered microemulsions or highly ordered self-assembled patterns. Similar phenomena occur not only in polymeric materials (block copolymer melts or .polymersomes. in solution), but also in mixtures of biological lipids and water.5
Due to this universality of the qualitative behavior, coarse-grained models are well suited to investigate these complex fluids. Coarse-grained models, which do not explicitly incorporate the structure on atomistic scales but only include the relevant interactions necessary to bring about the phenomena of interest, are able to bridge the pertinent length scale from a tens of nanometers to a micrometer and times scales from a nanosecond to a second.
Much effort has been directed towards mapping the structure and dynamics of chemically realistic polymers6 onto coarse-grained models, to devise simulation methods to extract free energies and to predict the behavior of spatially inhomogeneous systems.1–3 Interesting future topics include among others: glass transition of polymers in confinement,7 dynamics of soft matter at interfaces, computational methods for predicting structure formation and self-assembly in multi-component systems,8 coarse-grained models for biological mem-branes.5

FIG. 1: Bending-unbending transition of the adsorbed polymer layer on a striped polymer brush. The figure presents the probability distribution of the total film thickness Σ, a function of the fraction of the polymer brush x (cf. sketch in the inset). Around x ≈ 0.27 there is a morphological transition. The dashed lines mark the adsorption on the bare substrate, on the brush and the linear superposition. The panels on the right present snapshots of configurations at x =0.266. The simulation box and three periodic images are shown. The upper and lower panels correspond to Re2 / Σ = 1.38 and 5.67, respectively. From: M. Muller, L. Gonzalez MacDowell, J. Phys.: Condens. Matter 15, R609–653 (2003).
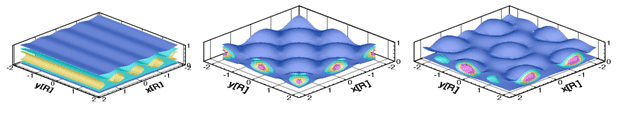
FIG. 2: Morphology of a mixed AB polymer brush. Grafting incompatible polymers A and B immobily onto a substrate prevents macrophase separation and the molecules arrange into two dimensional, periodic microdomains. Contours of the total density at Φ= 1/2 in the vicinity of the triple point, where a ripple-phase (left) coexists with a checkerboard structure (middle) and a hexagonal dimple phase (right). The segregated regions are more dense than regions where the A and B component mix. In the “ripple” phase (left) the species cluster into cylinders, every second one is rich in the A component. In the checkerboard phase(middle) A and B clusters alternate on a quadratic lattice. In the “dimple A” phase (right), the A component clusters and the dense regions form an hexagonal lattice, while the B component is collapsed and lls the space between the A-rich clusters. From: M. M�ller, Phys. Rev. E 65, 030802(R) (2002).
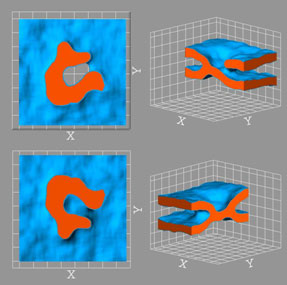
FIG. 3: Observed fusion intermediate in a coarse-grained model for lipid membranes. Two tense membranes in apposition spontaneously fuse, but the fusion pathway is not axially symmetric but involves the formation of a pore in a membrane close to the initial contact (stalk) of the two membranes. Fusion is spatially and temporally correlated with transient leakage. Four different views of the intermediate are presented. Hydrophobic core is shown as dark gray, the hydrophilic-hydrophobic interface (defined as a surface on which densities of hydrophilic and hydrophobic segments are equal) is light gray. Hydrophilic segments are not shown for clarity. From: M. M�ller, K. Katsov, M. Schick, J. Polyn. Sci. B21: Polym. Phys. 1441–1450 (2003).
Abstract: One of the most compelling frontiers for materials scientists is to be found in the living world. Recent advances in molecular biology, x-ray crystallography and electron tomography and single molecule biophysics have opened up possibilities ranging from synthetic proteins to devices and sensors inspired by their cellular hosts. The aim of this talk will be to describe some of the critical modeling challenges that must be faced in considering macromolecules and their complexes. I will feature two case studies, one involving the elasticity of DNA and its relevance to the packing of DNA in viruses and eucaryotic cells and the second that considers the physical mechanisms of mechanosensation. These case studies will highlight both the power and shortcomings of both atomistic and continuum descriptions of biological problems and will illustrate the important unanswered challenges in this area.
There is now a considerable effort to foster cooperative research efforts between groups though collaborative networks, both in the European Union and in the US. For example the Psi-k network in the EU has been particularly effective in advancing theories and algorithms for electronic structure calculations. In the US, the much newer Computational Materials Science Network (CMSN) of the Department of Energy Basic Energy Sciences [see http://www.phys.washington.edu/~cmsn/] is beginning to make a comparable impact. Such networking is particularly important in computational materials science, where code development takes years of dedicated work, and research is becoming more and more dependent on modern codes such as VASP, GAUSSIAN, WIEN2K, in addition to specialized codes developed by a single group. Currently the CMSN supports five major projects, each representing a major scientific problem of interest to various disciplines, such as magnetism and magnetic materials. Each of the projects typically links a dozen or more teams of researchers, using a combination of travel, workshops and shared postdocs. Although the funding currently provides only modest supplements to existing research grants, the closer cooperation spurs research significantly, leading to developments that could not have been achieved so quickly with independent efforts.
Thus it seems clear that linking US and EU networks would have many comparable advantages. There are several fields of research where the separate efforts of US and EU researches could benefit from more frequent collaborations. For example, there is considerable interest both in the US and the EU in emerging field of excited state electronic structure. Progress in this field requires theoretical treatments that go beyond the effective independent particle of ground state density functional theory, and various theories are under active development. For example a first principles treatment of excited states demands a computationally demanding two particle theory such as the Bethe-Salpeter equation, and hence points to the need for more efficient approaches such as the time dependent density functional theory (TDDFT). Given the common interest in this field, efforts are beginning to be made to foster more cooperation. For example, this year some key representatives of US research teams will be invited to the EU NANOEXE2004 workshop in September 2004.
There is also a serious need to link various computer codes more seamlessly. For example, it would be desirable to link Car-Parrinello MD codes with excited state codes to take advantage of the capabilities of each. Likewise, it would be useful to link the WIEN2K all electron code with excited state codes like FEFF8 for spectroscopy calculations. Such linking would avoid needless duplication of effort, and would enable many research group to share improved state-of-the-art codes. Indeed, an effort to improve code sharing would be beneficial to a large number of research groups both in the US and the EU. This would also bring together several research groups interested in serious code development.
Kinetic Monte Carlo simulation is a useful tool to study the dynamics of physical and chemical systems on mesoscopic and macroscopic time scales much longer than the picosecond scales accessible with molecular dynamics. However, the Monte Carlo transition rates are not, in general, known from first principles. Often it is therefore assumed that 'minor' differences between dynamics are not very important, as long as they obey detailed balance and thus eventually bring the system to thermodynamic equilibrium. In this talk I will show that this view is too simplistic, and that very significant differences are found in the structure and mobility of driven interfaces,1 as well as in nucleation rates at low temperatures,2 between kinetic Ising systems evolving under different stochastic dynamics. In particular, I will discuss the differences between "hard" dynamics (such as the standard Glauber and Metropolis rates), in which the effects of the interactions and the applied field do not factorize in the transition rate, and "soft" dynamics that possess such a factorization property. In addition to the hard and soft Glauber cases, I will also consider rates that contain local energy barriers between the individual system states,3 including the one-step-dynamic4 and the transitiondynamics- approximation.5 The moral of my story is that great care must be shown in devising stochastic Monte Carlo dynamics for specific systems if the time-dependent results are going to be physically meaningful.
References
Kinetic Monte Carlo (KMC) simulations provide one of the very few methods to extend simulations of dynamic phenomena from the picosecond scale of microscopic phenomena to the scales of seconds to millions of years that are of interest in macroscopic scientific and engineering applications. An illustration of such extreme long-time simulations is given in Fig. 1, which shows the average lifetime of the metastable magnetization state in a three-dimensional Ising model evolving under the Glauber dynamic1 at a temperature of 0.6Tc (measured in units of Monte Carlo steps per site, MCSS).2 This simulation was performed with the projective-dynamics method with a moving wall.3 To put these extreme timescales in perspective, one might mention that the age of the Universe, measured in femtoseconds, is about 1032.
However spectacular the timescales accessible with KMC might be, the method is still plagued by two fundamental problems. First, the basic Monte Carlo attempt frequency (or its inverse, the basic time constant) must be obtained by comparison, either with more microscopic simulational or theoretical methods, such as molecular dynamics, possibly in combination with quantum mechanics, or with experiments. Second, in order for the dynamical results of the simulation to be meaningful, the Monte Carlo transition rates must be appropriate for the specific physical or chemical system under consideration. Here, I will concentrate on the second of these problems.
In equilibrium Monte Carlo simulations one is only concerned with sampling equilibrium configurations as effectively and efficiently as possible, and so there is no concern about whether or not the specific sequence of configurations corresponds to the true time evolution of the systems. Thus, the only requirement that the rates must satisfy is detailed balance. Often used transition rates are those of Metropolis et al.4 or Glauber,1 and in the past it has often been assumed that the differences between the dynamical behaviors of systems governed by different dynamics are only minor, as long as detailed balance is obeyed and conservation laws are not changed. However, it has recently been demonstrated that even seemingly minor differences can have remarkably strong effects. One example is the difference between .hard. dynamics, in which the effects on the transition rates of interaction energies and fields (or chemical potentials) do not factorize, and “soft” dynamics, in which they do.5 While the hard dynamics cause considerable kinetic roughening of interfaces driven by an applied field or chemical-potential difference, such roughening is all but absent for soft dynamics. As a result, the interface mobility with hard dynamics is considerably larger than with soft dynamics.6 Recently it has also been shown that low-temperature nucleation under soft dynamics remains an activated process, even for very strong fields. This is not the case for hard dynamics (see Fig. 2).7 Differences in nucleation rates have also been demonstrated between popular KMC rates that include local energy barriers between individual states, such as the one-step dynamic8 and the transition-dynamics algorithm.9 However, these differences are not as spectacular as those between the hard and soft Glauber dynamics.10
A conclusion one can draw from the results discussed above is that great caution must be exercised when developing transition rates for KMC studies of specific systems. Some examples on how this can be done using a density-matrix formalism are given in Refs.11, 12 In11 it is shown that the standard Glauber dynamic can be obtained from a system of S=1/2 particles weakly coupled to a fermion bath, while a new dynamic, in which transitions between states of the same energy are forbidden, is derived in12 for S=1/2 particles weakly coupled to a boson (phonon) bath. Much more work in this direction is needed and will likely involve large-scale quantum-mechanical calculations requiring international collaborations.

Figure 1. The lifetime of the metastable magnetization state of a 163 Ising ferromagnet with Glauber dynamics at a temperature of 0.6Tc, measured in Monte Carlo steps per site (MCSS). The age of the universe, measured in femtoseconds, is approximately 1032.

Figure 2. Effective energy barrier ⋅ for low-temperature nucleation in a two-dimensional kinetic Ising model evolving under hard and soft Glauber dynamics, respectively. Nucleation with the soft dynamic remains activated in the strong-field limit. Inset: temperature times ln of the metastable lifetime, shown vs temperature. The zero-temperature limits of these lines give the values of ⋅ shown.
Interfacial processes dominate all friction, adhesion and nanoscale flows. However, the conditions at the interfaces are determined by deformation and flow at much larger scales. The challenge is to integrate an atomic-scale treatment of bond breaking, sliding, and flows at interfaces, with large scale information that is most efficiently described with continuum algorithms. This talk will briefly highlight several different approaches being used in our group. The first example is a study of craze formation and the fracture energy of glassy polymers. Here, atomistic simulations are used to study different phases of deformation and fracture. They provide information about the fundamental processes of craze formation, as well as constitutive relations that can be used in continuum calculations of the fracture energy. The second example is a mesoscale model for immiscible fluids. Most previous models were chosen for simplicity or convenience. We have fit detailed molecular dynamics (MD) simulations to a mesoscale model and shown that it reproduces a wide range of behavior that is not included in the fit. The fit reveals several fundamental shortcomings of previous models. The final example is simultaneous multiscale modeling where different regions of space are described with different levels of detail. An algorithm for fluids has been applied to slip at rough walls and singular corner flows in cavities. A parallel algorithm for solids has been applied to stick-slip motion and contact of rough surfaces.
In this contribution, hierarchical modeling aspects are presented for the system Fe-Cu in order to simulate strengthening effects due to Cu-precipitates appearing under thermal loading conditions: The Monte Carlo method is employed to simulate the growth of Cu-precipitates during long time annealing at moderate temperatures. Dislocation theory and Molecular Dynamics are used to understand the dislocation/precipitate interaction and the strengthening related with these precipitates. Finally, the macroscopic behavior is analyxed with the Rousselier damage mechanical approach. The analysis is extended to multiphase precipitates containing Ni and Mn in addition to Cu. Their composition is obtained experimentally as well as numerically and both are found to be in good agreement. In addition, atomistic and micromechanical numerical analyses are presented on the mechanical behavior and damage behavior of different particle and fiber reinforced composites using cell models, self-consistent models and matricity models as well as for hydrogen embrittlement and P and C diffusion on lattice and interstitial lattice positions in Fe. The results are shown to be in good agreement with experiments. Key issues for future investigations are derived and the benefit from possible co-operations with US-researchers in theses fields are discussed.
Key Issues in the Field:
Advance of the Field by US-EU collaboration:

Relevant lengths scales and approaches in materials modeling
Example of microscopic-macroscopic coupling et the continuum level
References
The talk will cover numerical methods and their application to bridge the time and length scale for the simulation of ultra-high density future magnetic storage systems. Examples will be given1 For the estimation of the failure rate of magnetic random access memories calculating the relevant energy barriers.2 For the simulation of magnetization processes in magneto-electronic field sensors describing the physics on a mesoscopic length scale.3 For the simulation of the read and write process in magnetic recording using fast boundary methods to bridge the length scale.
Recent References
Current Activities Related to the workshop:
Bridging of length scales: Fully integrated recording simulations
With increasing magnetic storage density the overall dimensions of the hard disk systems become smaller. The full interactions between head and media must be taken into account to give a valid description of the magnetic recording process. A fully integrated recording model that uses the finite element model together with an H-matrix accelerated boundary element method. The so called hierarchical matrices (H-matrices) are a novel way to reduce CPU-time and memory requirements for long-range interactions. This allows the bridging of length scales towards large system size.

Bridging the length scale in recording simulations. Finite element model of the recording head showing the coil, the soft magnetic yoke, and the gap region. The size of the ring head spans several micrometers. (left hand side). 3D view of the data layer after recording four transitions. The grey scale maps the magnetization parallel to the down track direction. The bit width is 50 nm (right hand side).
Bridging of time scales: Long term stability of Magnetic Random Access Memories
With decreasing size of magnetic storage elements, thermal fluctuations deteriorate the life time of the information. The energy barrier between stable configurations determines the probability of a thermal switching event. In Magnetic Random Access Memory (MRAM) elements the shape of the element ("shape anisotropy") and the induced crystalline anisotropy determine the energy barrier. This should guarantee a lifetime of a stored bit of about 10 years. The nudged elastic band method finds minimum energy paths in high dimensional energy landscapes and is a rigorous way to compute the saddle point(s) between local energy minima.
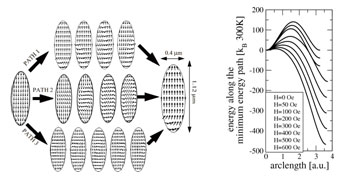
Bridging the time scales. The probability of rare events can be calculated from the saddle point energy. Thermally induced reversal modes of MRAM cells for different external fields (left hand side). Energy along the minimum energy path is illustrated for different applied fields (right hand side).
Key issues for US / EU collaboration
The development of advanced magnetic materials requires a precise understanding of the magnetic behavior. Prominent examples are magnetic recording systems for future high density information storage, where both recording medium and recording heads have to meet certain characteristics. These structures are so small that quantum mechanical effects like exchange have to be taken into account. However, they are too large for a pure quantum mechanical description, which would exceed the capabilities of today's ab initio computational models. On this intermediate level between the macroscopic world and a description with atomic resolution, micromagnetic models provide a useful tool for detailed predictions of the magnetic properties and magnetization processes.
Future activities will focus on the bridging of length scales in the field for the simulation of magneto-electronic devices and magnetic materials. Meso-scale simulations for the design and optimization of magnetic materials require detailed parameters, which can be only provided by ab initio simulations.
A joint work program will combine the complementary approaches of US and EU researchers. Examples of current activities show the EU and US expertise in computational magnetism:
Benefits from collaborative research are expected in the following areas:
Collaborative projects will enable the fully integrated simulation of magnetic devices including a realistic model of the magnetic materials involved. The simulation of the functional behavior takes into account the physics at different length scales, starting from the structural properties of the magnets and leading to the macroscopic properties of the device. The simulations will provide the theoretical background for the optimization and design of magnetic devices (magneto-electronic sensors, integrated read/write head for magnetic data storage, ultra high density magnetic storage media).
To examine fundamental mechanisms associated with size effects in plasticity, we perform simulation studies of dislocation dynamics in two dimensions. While highly idealized, these model systems provide insight into forces driving dislocation patterning and the emergence of characteristic length scales in plastic deformation. To study the dynamics and patterning of screw dislocations in two dimensions, we consider an idealized crystal deformed with a two-dimensional anti-plane strain field z(x,y). A close analog of the 2-d XY model from statistical physics, this system can contain point screw dislocations with Burgers vector in/out of the plane, but no edge dislocations. We simulate the dynamics of the strain field, and our simulation methodology allows us to study dislocation nucleation, motion, and annihilation without explicitly calculating the interactions and trajectories of individual defects. In a bulk system under a constant applied shear strain rate, we observe the coalescence of dislocation-rich slip bands whose spacing distribution obeys a simple scaling law. We then study plastic deformation under a strain gradient in the fiber pull-out and channel flow geometries, and in the presence of a purely ductile crack loaded in mode III; in each case we vary the system size and find clear evidence of size effects. A careful analysis of the system's constitutive behavior in the channel flow geometry shows that while most of the system obeys the Bingham plastic law, a surface layer emerges whose dislocation density is lower than expected and whose yield stress is higher than that of the bulk. We argue that dislocation image interactions near the boundary force the Bingham plastic law inevitably to break down, so that the sample's mechanical response is always harder near the surface than in the bulk. Next we study the dynamics and patterning of edge dislocations under a strain gradient in two dimensions, by examining the response of a two-dimensional crystal under bending using both single crystalline and polycrystalline initial states. The system evolves via a Monte Carlo algorithm to approximate quasi-static loading at finite temperature. Deformation arises from nucleation and motion of edge dislocations. We observe the onset of spontaneous dislocation patterning and the nucleation and growths of well defined tilt boundaries, and examine both size effects and strain rate effects in this system. Lastly, we discuss ways in which statistical mechanics may provide useful insights into the mechanisms driving dislocation patterning and the emergence of characteristic length scales in plastic deformation. This work was supported by the National Science Foundation under grant DMR- 0116090 and by the NIST Center for Theoretical/Computational Materials Science.
The sub 10nm length scale of CMOS devices creates new materials challenges for which modeling of materials, interfaces, dopants, and defects becomes crucial to process design and understanding device performance. The critical integrated circuit components will comprise tens of thousands of atoms or less, so approaching molecular scales. The widely-recognized intrinsic parameter fluctuations from discreteness of charge and matter will be a major factor limiting scaling and integration. Despite some progress in understanding these phenomena, there are no adequate models or simulation tools able to predict accurately the origins, characteristics, scale and consequences of the intrinsic fluctuations in such molecular-size devices. I will discuss two interrelated issues: i) how to bridge the gap between materials modeling and device simulation, and ii) how modeling of Atomic Force Microscopy (AFM) may help to study defects in oxides and manipulate molecules to produce molecular devices. Both represent applications of multiscale computational approach to modeling complex processes at surfaces and interfaces of wide-gap insulators. I will outline challenges related to disorder and statistical nature of defect states in these nano- scale devices and demonstrate how one can use an embedded cluster method to tackle some of these issues. Recent experiments using noncontact AFM demonstrate that the short-range interaction forces can be measured selectively above chemically identified sites on surfaces of insulators. These experiments and atomically resolved AFM imaging of insulators rely on multi-scale theoretical models for their interpretation. I will discuss the applications of AFM modeling to study the structure and spectroscopic properties of surface point defects and adsorbed molecules and for molecular manipulation.
Challenges: dimensions (less than 100 dopant atoms)and defects (a single defect in the oxide can break a device)

Snapshot from the molecular dynamics simulations of a CF2 fragment bonding to polystyrene chains. C in the CF2 is shown in green, C in the polystyrene is shown in blue, F is shown in red, and H is shown in white.
Fluorocarbon plasmas are widely used to chemically modify surfaces and deposit thin films. It is well-accepted that polyatomic ions and neutrals within low-energy plasmas have a significant effect on the surface chemistry induced by the plasma. For this reason, the deposition of mass selected fluorocarbon ions are useful for isolating the effects specific to polyatomic ions. In this study, the detailed chemical modifications that result from of the deposition of beams of polyatomic fluorocarbon ions (C3F5+ and CF3+) on polystyrene surfaces at experimental fluxes are identified using classical molecular dynamics simulations with many-body empirical potentials. To facilitate these simulations, a new CH-F potential is developed based on the second generation reactive empirical bond-order potential for hydrocarbons developed by Brenner. Lennard- Jones potentials are used to model long-range van der Waals interactions. Both beams are deposited normal to the surface at incident energies of 50 eV/ion. For CF3+ deposition, F atoms play the most important role in fluorinating the polystyrene surface, as the majority of them are covalently attached to the polymer chains through replacement of native H atoms or capping the end of broken chains. CF2 fragments are also an important long lived species. In contrast, F atoms are a minor bi-product and CF2 fragments are the most dominant species for C3F5+ deposition on polystyrene. Thus the simulations explain the experimental finding that C3F5+ is more efficient at producing fluorocarbon thin films. In particular, many larger fragments produced by C3F5+ ion deposition, such as CF2, C2Fn and C3F5, contain more than one C atom and may have more than one active site. These larger fragments readily react and connect with other fluorocarbon ions or fragments to grow polymer-like structures, as illustrated in the figure. In contrast, F atoms, the most dominant fragment in CF3+ deposition, effectively deactivate potential film nucleation sites when they fluorinate the polymer surface. These findings can be generalized to state that larger polyatomic ions produce a wider variety of precursors for film growth than smaller polyatomic ions and thus should be more effective at growing thin films from plasma. Additionally, they suggest that CF2 is an important precursor for film growth from fluorocarbon polyatomic ions. This work is supported by the National Science Foundation (CHE-0200838).
Key Problems:
Extending simulations to longer times by combining MD and MC simulations, accelerated MD
Developing improved methodologies for modeling chemistry on large scales: embedding tight-binding or QM methods in empirical potentials
How US/EC Collaborations Would Help
Enhanced collaboration between computational and experimental groups, especially in the area of plasma-surface modification, would enhance validation of computational methods. The US has strong expertise in fluorocarbon (FC) modeling while the EC has strong expertise in experimental FC plasma deposition of polymer films.
US and EC have complementary expertise in computational modeling, especially in mesoscale modeling and accelerated dynamics on the EC side, and potential development and empirical classical dynamics on the US side, that should be combined to better solve problems.
Snapshot from a classical MD simulation of CF2 (shown in green and red) bonding to PS chains after polyatomic ion beam deposition.

Snapshot from classical MD simulations of FC oligomers formed from C3F5+ deposition at incident energies of 50 eV/ion.
References
“Effects of Unique Ion Chemistry on Thin-Film Growth by Plasma-Surface Interactions,” M.B.J. Wijesundara, L. Hanley, B. Ni and S.B. Sinnott, Proceedings of the National Academy of Science, USA 97 23-27 (2000).
“Quantifying the Effect of Polyatomic Ion Structure on Thin-Film Growth: Experiment and Molecular Dynamics Simulations,” M.B.J. Wijesundara, Y. Ji, B. Ni, S.B. Sinnott, and L. Hanley, Journal of Applied Physics 88, 5004–5016 (2000).
“The Growth and Modification of Materials via Low Energy Ion-Surface Processing,” L. Hanley and S.B. Sinnott, Surface Science 500, 500–522 (2002).
“Effect of Surface Structure on the Results of C3H5+ Deposition on Polymers: Predictions from Molecular Dynamics Simulations,” I. Jang, R. Phillips and S.B. Sinnott, Journal of Applied Physics 92, 3363–3367 (2002).
“Molecular Dynamics Simulations of Thin Film Nucleation through Molecular Cluster Beam Deposition: Effect of Incident Angle,” Y. Hu and S.B. Sinnott, Nuclear Instruments and Methods in Physics Research B 195, 329–338 (2002).
“Second Generation Reactive Empirical Bond Order (REBO) Potential Energy Expression for Hydrocarbons,” D.W. Brenner, O.A. Shenderova, J.A. Harrison, S.J. Stewart, B. Ni, S.B. Sinnott, Journal of Physics: Condensed Matter 14, 783–802 (2002).
Spin-polarized electronics is a rapidly expanding research area, both because of the fascinating fundamental physics observed in new spintronic materials, and because of their potentially far-reaching technological applications. Here we illustrate the utility of modern computational methods in the design and optimization of new spintronic systems by describing the successful prediction and subsequent synthesis of a new “multiferroic” material (which is simultaneously ferromagnetic and ferroelectric). Finally we mention some recent advances in computational methods that have allowed us both to understand the novel phenomena observed in spintronic materials, and to design improved materials for specific technological applications.
Computational design of multifunctional materials.
The areas in which computational methods can contribute to the design of new multifunctional materials are:1
The most widely-used tool for first-principles prediction of new materials is density functional theory (DFT). Examples of new material classes that have been successfully predicted using DFT include the half-metallic antiferromagnets (Fig. 1)2 and ferromagnetic ferroelectrics (Fig. 2).3
There are two main challenges. The first is the development and implementation of exchange-correlation functionals that are physically appropriate and computationally affordable. Many multifunctional materials of interest fall into the category of "strongly-correlated" materials, in which the substantial interactions between the electrons render traditional methods such as the local density approximation inadequate. Here some progress has been made with, for example, the LDA+U4 and pseudodpotential self-interaction corrected (pseudo-SIC)5 methods, but more work is clearly needed. Second, many multifunctional materials consist of complex building blocks made of large numbers of atoms, and contain defects such as vacancies or dopants that strongly influence the properties. Our ability to compute the properties of such large complex systems depends on the continued development of improved computational algorithms, such as order-N methods, or parallel implementations. In both cases, collaboration with European groups who maintain large, publicly available density functional codes (such as ABINIT, VASP and SIESTA) will be particularly productive.
References
Isocontour plot of the magnetization density of the HM AF state calculated for La2VCuO6. The red-yellow surface surrounds the Cu2+ ion, while the larger blue surface surrounds the V4+ ion; the spin directions are opposite but the magnetization of the spin densities are the same on these surfaces. The other blue surfaces illustrate the polarization of oxygen 2p states, which is in the same direction as the Cu spin. The color is determined by the magnitude of the gradient of the spin density, not by the direction of spin. Two of the double perovskite unit cells are pictured. From Ref.2

Isosurface (at a value of 0.75) of the valence electron localization function of BiMnO3, calculated at its experimental structure. The projection on the back face of the cell shows the valence electron localization function, color-coded as in the bar by the side of the figure. The crystal is oriented slightly off the b axis. From Ref.3
A significant problem in the atomistic simulation of materials is that molecular dynamics simulations are limited to nanoseconds, while important reactions and diffusive event often occur on time scales of microseconds and longer. Although rate constants for slow events can be computed directly using transition state theory (with dynamical corrections, if desired, to give exact rates), this requires first knowing the transition state. Often, however, we cannot even guess what events will occur. For example, in vapor-deposited metallic surface growth, surprisingly complicated exchange events are pervasive. I will discuss recently developed methods (hyperdynamics, parallel replica dynamics, and temperature accelerated dynamics) for treating this problem of complex, infrequent-event processes. The idea is to directly accelerate the dynamics to achieve longer times without prior knowledge of the available reaction paths. Time permitting, I will present our latest method developments and some recent applications, including metallic surface growth, deformation and dynamics of carbon nanotubes, and annealing after radiation damage events in MgO.
The desire to miniaturize metallic components for application to, for example, MEMS devices has revealed the existence of three types of size effects concerning the mechanical properties: the first is introduced by the measurement technique, the second by an interface dominated microstructure and the third by the reduced physical dimensions of the metallic component.
A path towards developing an understanding of these size effects is by exploiting the synergies between simulation and experiments. In this contribution it will be shown by means of a number of examples how simulations can help in the understanding of experimental results and how they can also form the basis for the development of new experimental techniques.
One of the best examples where such a synergy is applied, is the search for understanding the deformation mechanism in nanocrystalline metals. The main question is whether dislocation activity can still be considered as an important deformation mechanism in nanocrystalline metals with a mean grain size below 100nm. The question is difficult to answer since direct visualization techniques such as transmission electron microscopy (TEM) fail to give a clear answer.1 Post-mortem analysis revealed no dislocation debris but also no evidence for a grain boundary sliding mechanism. In-situ deformation in TEM demonstrates dislocation activity, but these experiments have two drawbacks. Firstly, one always follows the propagation of a crack tip that is known to possibly activate different deformation mechanisms, and secondly, TEM techniques suffer from a thin film approach.
The simulations have revealed three deformation modes in tensile deformation for samples with defect-free grain sizes below 30nm: the first identified as GB sliding governed by atomic shuffling and to some extent stress assisted free volume migration,2–4 and the second, gaining in importance at larger grain sizes, identified by a dislocation mechanism where GBs operate as a source and sink for lattice dislocations; a process through which the remaining GB dislocations are continuously redistributed.5 Both processes are intimately linked to GB and triple junction migration resulting in collective grain activity leading to the formation of mesoscopic shear planes that explains the dimple-like features on the fracture surface.6 The dislocation activity is mediated by partial dislocations: in some fcc metals such as Ni and Cu, the partials extend throughout the entire grain, whereas in material such as Al, a trailing partial is emitted soon after the leading partial, resulting in a full dislocation that travels through the grain.7 It has however been demonstrated recently that the extended partial dislocations might be a high stress/short time artifact of the MD simulation and that full dislocations have to be expected when extrapolating the simulation results towards experiments.8 In other words, this means that the deformation mechanism in nanocrystalline metals is still based on dislocations, but in this case the dislocations are emitted from and absorbed in the grain boundary, thus leaving no dislocation debris.
The suggestions from molecular dynamics have inspired us to develop a new in-situ technique, based on well known peak profile analysis methods, for addressing the relationship between microstructure and mechanical properties in nanostructured materials. It is well known that the dislocation network built up during deformation of polycrystalline metals result in an irreversible broadening of the diffraction peak. In-situ deformation measurements on electrodeposited Ni with a mean grain size of 26nm showed that the peak broadening is totally reversible upon unloading, demonstrating that no permanent dislocation debris is built up, as was predicted by the atomistic simulations.9 It will be shown however that not all materials classified under nanocrystalline metals show this reversible peak broadening and that the new in-situ technique can also be applied for the investigation of size effects in thin films.
References